

Abstract
Objective. Our 24-week study (NCT01197755; OSKIRA-3) compared the efficacy and safety of fostamatinib versus placebo in patients taking background methotrexate treatment with active rheumatoid arthritis (RA) and an inadequate response to a single tumor necrosis factor-α antagonist.
Methods. Adult patients were randomized (1:1:1) to fostamatinib [100 mg bid for 24 weeks (n = 105; Group A)], or 100 mg bid for 4 weeks, then 150 mg qd (n = 108; Group B), or to placebo (n = 110; Group C) for 24 weeks. Nonresponders at Week 12 could enter a longterm extension study. The primary endpoint was the proportion of patients achieving an American College of Rheumatology 20% (ACR20) response at Week 24.
Results. Baseline characteristics were well balanced. Significantly more patients in fostamatinib Group A (36.2%; p = 0.004), but not B (27.8%; p = 0.168), achieved ACR20 at Week 24 versus placebo (21.1%). Frequently reported adverse events were diarrhea, hypertension, and headache. Elevated blood pressure (≥ 140/90 mm Hg) at ≥ 1 visit was observed in 46.7%, 51.9%, and 26.6% of patients, respectively. There were 2 deaths in the study, 1 in Group B and 1 in the placebo group.
Conclusion. Fostamatinib 100 mg bid, but not fostamatinib 100 mg bid for 4 weeks then 150 mg qd, achieved statistical improvements in ACR20 at 24 weeks versus placebo. Because of efficacy and safety results from the phase III clinical program, the companies developing fostamatinib have decided not to study it further in RA at this time.
Spleen tyrosine kinase (SYK) has a central role in cell signaling in multiple immune cell types involved in the inflammation and tissue damage characteristic of rheumatoid arthritis (RA)1,2. SYK activation is implicated in the production of inflammatory cytokines and metalloproteinase3. Fostamatinib is an oral SYK inhibitor that has been investigated for the treatment of RA4,5. It is a prodrug that is rapidly and extensively converted to the active metabolite R406 by gut enzymes6, while the active metabolite blocks SYK-dependent immune-cell activation mediated by the immunoreceptors FcɛR, FcγR, and BCR. Therefore, inhibition of SYK by fostamatinib may reduce or prevent tissue damage associated with the disease.
Two phase II clinical studies (TASKi1 and TASKi2) in patients with RA who were methotrexate (MTX) treatment failures demonstrated that fostamatinib produced significant improvements in the signs and symptoms of RA when taken in combination with MTX6,7. Meanwhile, a third phase II clinical study (TASKi3) carried out in patients who did not respond to biologic agents (excluding MTX) found no statistically significant difference in the primary endpoint [American College of Rheumatology 20% (ACR20) response rates at Week 12] between fostamatinib-treated patients and those taking placebo (both in combination with MTX)8. An unusually high placebo response rate and unbalanced patient baseline characteristics may have contributed to this result8.
This phase III, 24-week study (NCT01197755), part of a larger fostamatinib phase lll (OSKIRA) study program, aimed to evaluate the efficacy of 2 oral dosing regimens of fostamatinib taken in combination with MTX, compared to placebo plus MTX, in patients with active RA who had experienced an inadequate response to a single tumor necrosis factor-α (TNF-α) antagonist.
MATERIALS AND METHODS
Study population
Male and female patients aged ≥ 18 years with active RA and an inadequate response to a single TNF-α antagonist were recruited for our study. Patients diagnosed with RA after age 16 years were included if they were currently, or previously, receiving a single TNF-α antagonist and had a washout period as follows: etanercept, 4 weeks; adalimumab, certolizumab, infliximab, golimumab, 8 weeks. They also were included if they had been taking MTX for at least 6 months prior to randomization (stable dose between 7.5 and 25 mg per week for at least 6 weeks prior to randomization) and had ≥ 6 swollen joints and ≥ 6 tender/painful joints (from 28-joint count), with either an erythrocyte sedimentation rate (ESR) ≥ 28 mm/h or C-reactive protein (CRP) level ≥ 10 mg/l. Other inclusion criteria were evidence of at least 1 of the following: positive rheumatoid factor, radiographic erosion, or presence of serum anticyclic citrullinated peptide antibodies.
Subjects were excluded if they had poorly controlled hypertension [≥ 140 mm Hg systolic blood pressure (SBP) and/or ≥ 90 mm Hg diastolic blood pressure (DBP)], recent or significant cardiovascular disease, liver disease or significant liver function test abnormalities, significant active or recent infection (including tuberculosis, and defined as a positive serological test for hepatitis B or C; patients with suspected human immunodeficiency virus; treatment with intravenous antibiotics within 1 month prior to randomization; treatment with oral antibiotics within 2 weeks prior to randomization; or current evidence of a clinically significant active infection), severe renal impairment, or neutropenia. Patients were also excluded if they had previously been treated with biologic agents other than TNF-α antagonists.
Patients were randomized (1:1:1) to receive 1 of the 2 dosing regimens of oral fostamatinib or a matching placebo regimen, in combination with their MTX therapy (Figure 1). Group A received fostamatinib 100 mg twice daily (bid) for 24 weeks; Group B, fostamatinib 100 mg bid for 4 weeks, followed by once-daily (qd) dosing with fostamatinib 150 mg up to Week 24; and Group C, placebo bid for 24 weeks.
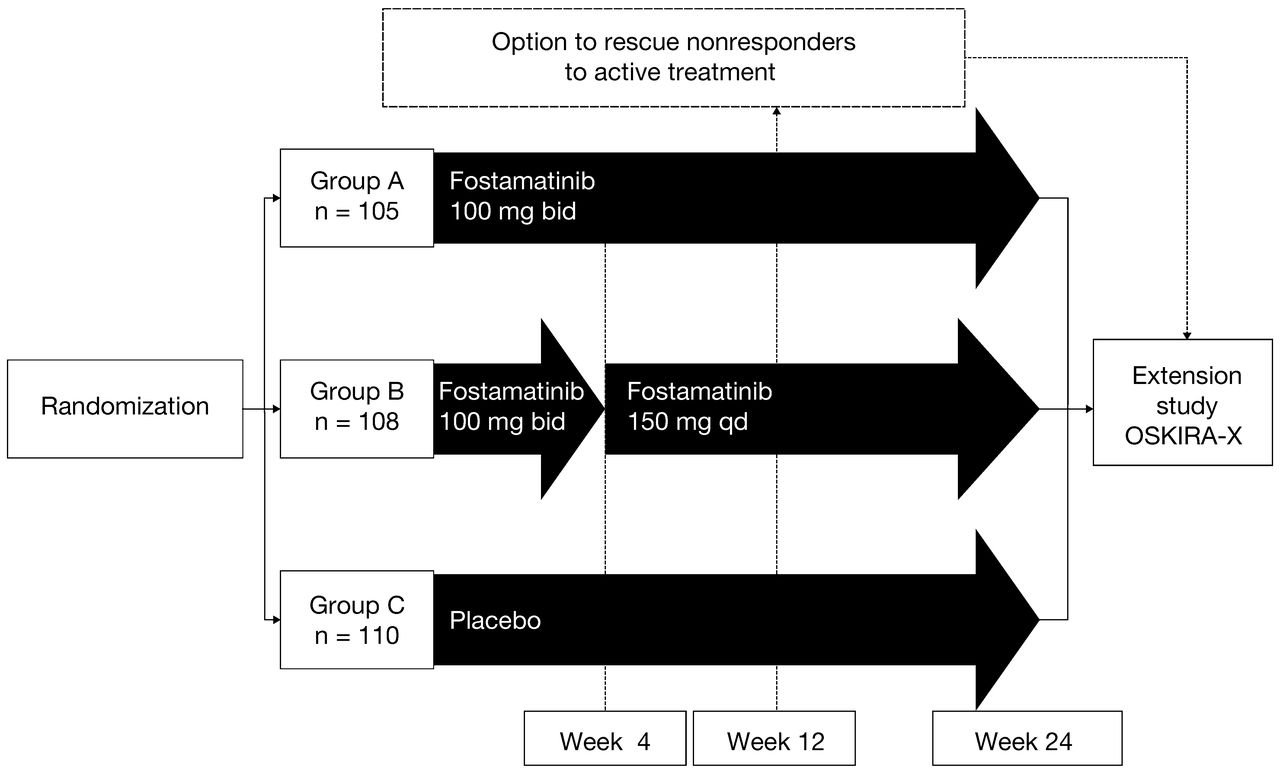
Study design. Randomized treatment given on background of methotrexate. Dose reductions in randomized treatment were permitted at any time in study provided certain criteria were met. bid: twice daily; qd: once daily.
If necessary, to control symptoms of RA, parenteral steroids were permitted during the study with the following restrictions: (1) 1 injection allowed between Week 4 and Week 16; (2) no injections allowed between Week 16 and Week 24, i.e., within 8 weeks of the primary outcome measures, to preserve the integrity of the results.
All treatment allocations remained double-blinded throughout the 24-week treatment period. Patients who successfully completed 24 weeks of treatment and those designated nonresponders at Week 12 (defined as not achieving at least a 20% reduction from baseline in either swollen or tender joint count) were offered the opportunity to receive fostamatinib therapy in a longterm followup extension study (OSKIRA-X, NCT01242514) while remaining blinded to OSKIRA-3 treatment.
Patients who entered the longterm extension study as nonresponders at Week 12 received 100 mg bid fostamatinib regardless of their original randomized dosing regimen, unless they had previously had a dose reduction. Patients who had their dose reduced to 100 mg qd continued at this reduced dose in the extension study. If those patients were originally allocated to the placebo group, they were switched to receive the reduced dose of 100 mg qd in the extension study (i.e., to match the dose-reduction fostamatinib regimen). Where dose reduction was required, the original treatment allocation remained blinded, although patients were aware of the switch to a reduced regimen. Data from the longterm extension study are not included in the results presented here.
Clinical efficacy outcomes
The primary outcome of our study was to assess the efficacy of fostamatinib in terms of the signs and symptoms of RA by measuring the ACR20 response rates at Week 249. Key secondary objectives of the study included measurement of ACR20 at Week 1, ACR 50% response criteria (ACR50), and ACR 70% response criteria (ACR70), assessment of physical function using the Health Assessment Questionnaire-Disability Index (HAQ-DI), and measurement of Disease Activity Score based on a 28-joint count (DAS28-CRP). Other secondary objectives included assessment of the prevention of structural joint damage, as measured by change in radiographic van der Heijde modified total Sharp score (mTSS) and the components of mTSS at Week 2410,11. In addition, consistency of results across a number of factors was evaluated for baseline HAQ-DI, DAS28-CRP, rheumatoid factor, duration of disease, sex, race, weight, age, and region. Posthoc subgroup analyses were also carried out for patients with erythrocyte sedimentation rate (ESR) ≥ 28 mm/h, ESR < 28 mm/h, CRP ≥ 10 mg/l, and CRP < 10 mg/l.
Safety outcomes
Safety outcomes included adverse events (AE), vital signs, electrocardiogram (ECG), clinical chemistry, hematology, urinalysis, and physical examination. Systemic drug levels of R406, the active metabolite of fostamatinib, were examined at weeks 4 and 24. The AE monitoring of unblinded data was carried out by an independent safety review committee with a blinded independent adjudication of cardiovascular events. If blood pressure (BP) exceeded 160 mm Hg SBP and/or 100 mm Hg DBP, a recommended protocol algorithm was used that included antihypertensive medication. Fostamatinib dose was reduced if BP remained persistently elevated.
Statistical methods
The study was initially designed to randomize about 450 patients. However, because of slower than anticipated recruitment, the planned total was revised to about 300 patients (Figure 1). A sample size of about 300 patients in total (100 in each arm) provided about 85% power to detect a 20% difference in the proportion of patients achieving an ACR20 response at Week 24 versus placebo.
The full analysis set was used as the primary analysis population for reporting efficacy, safety, and demography data. This comprised all patients randomized into the study who received at least 1 dose of study drug.
The primary endpoint was assessed for each dose regimen versus placebo using the Hochberg procedure12 to account for multiple testing, allowing for the correlation between the 2 comparisons because of the shared placebo data13 at the overall 2-sided 5% level. The primary analysis was performed by testing treatment difference in the proportion of ACR20 responders with a Mantel-Haenszel approach stratified by country. Based on the nonresponder imputation method, patients with no postbaseline data, who withdrew for any reason, had a disease-modifying antirheumatic drug initiated, or had an increased dose of background MTX, were considered nonresponders for ACR20 at all subsequent visits. Patients who received parenteral steroids had nonresponse imputed for 8 weeks following this event. Key secondary endpoints were assessed using the Hommel method of p-value adjustment for multiple hypothesis testing14. Safety data and tolerability data were assessed and summarized in terms of AE, ECG data, and changes in laboratory data, body weight, and vital signs.
RESULTS
Patient population
A total of 638 patients across 125 centers worldwide were screened for the study, of whom 315 did not meet the inclusion criteria. Overall, 323 patients were randomized, of whom 322 (99.7%) received at least 1 dose of investigational product. All patients randomized to Groups A and B received treatment (n = 105 and n = 108 patients, respectively). All but 1 of the patients randomized to Group C received placebo (n = 109). The patient was randomized but removed from the study for protocol noncompliance. Two patients from Group C were unblinded during the study at the investigator’s request; 1 because of a serious AE (severe anemia) and 1 following a sudden death (reported as from diabetes mellitus).
The treatment groups were well balanced demographically, although baseline mTSS was slightly higher in the fostamatinib groups versus placebo (Table 1). The majority of patients were recruited from North America (43.5%) or Latin America (35.7%). The baseline disease (RA) characteristics of the overall patient population were indicative of patients with RA who were inadequate responders to a TNF-α antagonist, with either erosive or seropositive disease (Table 1).
Demography and baseline disease characteristics: full analysis set.
Discontinuations
In total, 63.8% and 60.2% of patients in fostamatinib Groups A and B, respectively, completed treatment (including patients who had a dose reduction). Of the patients who received placebo, 50.0% completed treatment. The most common reasons for discontinuation of treatment were nonresponse at Week 12 (17.1%, 19.4%, and 31.8% of patients in Groups A, B, and C, respectively) and AE (7.6%, 9.3%, and 9.1% of patients, respectively; Table 2).
Patient disposition. A total of 638 patients were screened for the study, of whom 315 did not meet the inclusion criteria.
Primary efficacy endpoint at Week 24
Fostamatinib achieved statistically significant improvement in ACR20 response rate at Week 24 in fostamatinib Group A (100 mg bid) but not Group B [100 mg bid/150 mg qd; 36.2% (p = 0.004) and 27.8% (p = 0.168), respectively] compared with Group C (placebo; 21.1%; Figure 2). These findings were consistent across subgroups based on various baseline characteristics, including differences in region and disease profile (e.g., ESR ≥ 28 mm/h, ESR < 28 mm/h, CRP ≥ 10 mg/l, and CRP < 10 mg/l).

Signs and symptoms of rheumatoid arthritis at Week 24 (ACR20, ACR50, and ACR70). Nonresponder imputation has been applied following premature withdrawal, or increased dose of MTX or any DMARD initiation, or for 8 weeks following receipt of any parenteral steroids, or for patients with no postbaseline data. Group B ACR50 and ACR70 show nominal p values (formal statistical testing could not be carried out because of the hierarchical testing procedure and the failure of the primary endpoint for Group B).
*Compared to placebo. ACR20: American College of Rheumatology 20% response; ACR50: ACR 50% response; ACR70: ACR 70% response; bid: twice daily; DMARD: disease-modifying antirheumatic drug; qd: once daily; MTX: methotrexate.
Secondary efficacy endpoints up to Week 24
Fostamatinib achieved statistically significant improvements in ACR20 response rate at Week 1 (Groups A and B pooled) compared with Group C (placebo; Table 3). A significant effect was also seen with fostamatinib 100 mg bid therapy versus placebo over the 24-week period for the ACR50 and ACR70 response rates, in the reduction of HAQ-DI and in the number of patients achieving DAS28-CRP < 2.6 and DAS28-CRP ≤ 3.2 (Table 3). Formal statistical interpretation could not be done for Group B versus placebo because statistical significance was not seen for the primary endpoint. Fostamatinib did not show a difference in change in mTSS at 24 weeks for Group A (nominal p = 0.729) compared to placebo (Table 3).
Efficacy outcomes: full analysis set.
Safety
After 24 weeks, the total patient exposure to fostamatinib was 39.6 and 39.8 years for Groups A and B, respectively, compared to 35.6 patient-years’ exposure to placebo. Systemic drug levels of R406 over time were examined in a subset of patients. The concentrations observed were in the range achieved in the TASKi3 study (data not shown). Throughout the 24 weeks, AE occurred in 82.9% and 75.0% of patients in the fostamatinib groups (Groups A and B, respectively) and 71.6% of patients in Group C (Table 4). The majority were mild or moderate in intensity: 10.5% and 6.5% of patients in the fostamatinib groups and 11.9% of patients in the placebo group had at least 1 AE that was considered severe in intensity. Serious AE occurred in 6.7%, 6.5%, and 5.5% of patients, respectively. AE leading to discontinuation of treatment occurred in 9.5%, 10.2%, and 8.3% of patients, respectively. There were 2 deaths reported in the study, 1 (cardiorespiratory arrest) in fostamatinib Group B and 1 (diabetes mellitus and hyponatremia) in Group C (placebo).
Adverse event (AE) randomization to study end: full analysis set.
The incidence of AE leading to a dose reduction in fostamatinib Groups A and B was 7.6% and 5.6%, respectively. A lower rate (1.8% of patients) was reported for placebo. The most frequently reported AE in Groups A, B, and C were diarrhea (20.0%, 26.9%, and 6.4%), hypertension (13.3%, 13.9%, and 8.3%), and headache (7.6%, 8.3%, and 10.1%), respectively.
Elevated BP (≥ 140/90 mm Hg) at ≥ 1 visit was observed in 46.7%, 51.9%, and 26.6% of patients in Groups A, B, and C, respectively. Mean changes from baseline after 4 weeks of treatment were 5.1, 4.9, and −0.3 mm Hg for Groups A, B, and C. At Week 24, mean changes in SBP were −1.6, 3.7, and 1.5 mm Hg, respectively. Mean changes from baseline in DBP after 4 weeks of treatment were 3.9, 3.7, and −0.9 mm Hg. At Week 24, mean changes in DBP were 1.6, 3.5, and 0.3 mm Hg, respectively.
There were 3 adjudicated cardiovascular events in Group B (7.3/100 patient-years; cardiopulmonary arrest with fatal myocardial infarction; heart failure; syncope). One event in the placebo group was adjudicated as indeterminate (2.7/100 patient-years; sudden death, reported as diabetes mellitus). There was 1 malignancy (2.4/100 patient-years; renal cell carcinoma) in Group B.
Over the 24-week study, the incidence of infection was 42.9%, 28.7%, and 24.8% patients in Groups A, B, and C, respectively. The most common infections in the fostamatinib treatment arms were upper respiratory tract infections and bacterial infectious disorders. The incidence of serious infection events (SIE) was 3, 2, and 2 patients in Groups A, B, and C, respectively, with the most common being gastroenteritis. No patients randomized to fostamatinib experienced an SIE due to neutropenia (< 1.0 × 109/l). There were few patients with opportunistic infections and 1 case of herpes zoster, a non-serious event, in a patient in Group C. No cases of tuberculosis were reported. Both fostamatinib groups showed a dose-related decrease from baseline in leukocyte and neutrophil counts. In total, 4.8%, 3.8%, and 2.8% of patients in Groups A, B, and C experienced a neutrophil count < 1.5 × 109/l and 2 patients (1 in Group A and 1 in Group C) experienced an absolute neutrophil count ≥ 0.5 to < 1.0 × 109/l. No patients experienced a neutrophil count < 0.5 × 109/l.
Some differences were observed between treatment groups in the majority of clinical chemistry variables. None of the changes in lipid chemistry were considered clinically relevant. Alanine aminotransferase (ALT) increases from ≥ 3 to < 5 × upper limit of normal (ULN) were seen in 4 patients in Group A, 2 patients in Group B, and 1 patient in Group C. No patients in Groups A and B had ALT increases ≥ 5 × ULN. One patient in Group C had increased ALT ≥ 5 to < 10 × ULN. Aspartate aminotransferase increases from ≥ 3 to < 5 × ULN were seen in 2 patients in Group A, 2 patients in Group B, and 0 patients in Group C. There were no reported AE of hepatotoxicity/hepatocellular injury.
DISCUSSION
This phase III study evaluated the efficacy and safety of 2 oral dosing regimens of fostamatinib, taken in combination with MTX, compared to placebo plus MTX in patients with active RA who had experienced an inadequate response or intolerance to a single TNF-α antagonist.
Because of slow recruitment of patients for our study, the decision was made to reduce the sample size from about 450 randomized patients to about 300 randomized patients. The slow recruitment was due to the difficulty in enlisting patients who had only received a single prior TNF-α antagonist (in general, patients are administered multiple successive TNF-α antagonists in an attempt to find one that is efficacious). This reduction in sample size reduced the power from about 95% to 85% to detect a difference of 20% or more between active treatment and placebo in the percentage of patients achieving ACR20 at Week 24, and the ability to detect a minimum of 11% treatment effect, using a 2-sided test at the 2.5% level of significance. Subsequent investigation of the results suggested that the conclusions of the study would have been the same had the sample size remained as planned, e.g., in terms of the percentage of patients who achieved ACR20 at Week 24, the difference between treatments seen was 15.1% for the Group A versus placebo comparison, and 6.7% for the Group B versus placebo comparison. Without this sample size reduction, therefore, fostamatinib would have achieved statistically significant improvements in the primary variable (ACR20 response rate at 24 weeks) in Group A but not in Group B, compared to Group C.
The efficacy results showed that fostamatinib 100 mg bid was superior to placebo for the primary outcome variable ACR20 response rate at 24 weeks, although only a low number of patients achieved this outcome — lower than expected in this patient population. This finding differs from a phase II study8 of patients who had not responded to biologic agents, which showed no statistically significant difference in ACR20 response rate following administration of fostamatinib 100 mg bid versus placebo. Patients in our present study treated with fostamatinib 100 mg bid for 4 weeks and then fostamatinib 150 mg qd did not achieve clinically meaningful or statistically significant results versus placebo.
Results were generally consistent across the secondary measures of signs and symptoms and functional assessment; however, formal statistical comparisons could not be done for Group B versus Group C owing to nonsignificance of the primary endpoint for this comparison. In addition, a dose response was generally seen across the signs and symptoms endpoints. A limitation of the study, however, was that it was powered to detect a difference in the proportion of patients achieving an ACR20 response, and because multiplicity adjustment was used for the key secondary endpoints, it was not powered to detect differences between groups in radiographic endpoints. Fostamatinib 100 mg bid did not show a difference in mTSS at 24 weeks compared to placebo (nominal p = 0.729). Another limitation of our study was that patients had failed only a single TNF-α antagonist and the results may not be generalizable to the broader, more refractory population.
Safety and tolerability findings for fostamatinib were consistent with those previously observed in the TASKi phase II program6,7,8, in which fostamatinib was given on a background of MTX or other traditional disease-modifying antirheumatic drugs. Diarrhea was the most frequently reported AE, and was more frequently reported with fostamatinib than with placebo, although a dose relationship was not apparent. Most events of diarrhea resolved on treatment with minimal intervention. The data on decreases in neutrophil counts in this study confirm observations in earlier phase II fostamatinib studies, demonstrating no association between SIE and decreases in neutrophil count associated with fostamatinib6,7.
Also consistent with previous studies is the observed association between fostamatinib and an elevation in BP6,7,15,16,17. In those studies, elevated BP typically occurred early on, and was generally resolved through dose reduction or antihypertensive therapy. This elevation could be due, in part, to the off-target effects of fostamatinib. Indeed, animal model and in vitro studies have shown that fostamatinib increases vascular resistance, a consequence of impaired vasorelaxation resulting from reduced endothelial nitric oxide availability that could lead to increases in BP. The incidence of hypertension as an AE across the treatment groups was 13.3% and 13.9% in the fostamatinib groups (A and B) and 8.3% in the placebo group (C), with increases with fostamatinib evident as early as Week 1. The profile of elevated BP from baseline over time was more pronounced in Group A than Group B. Patients in all 3 groups were more likely to develop elevated BP if they were taking antihypertensive medication at baseline. The effect of fostamatinib on 24-hour ambulatory SBP in patients in the OSKIRA study program (the ambulatory BP monitoring trial) will be addressed in a separate publication.
In this phase III study in patients with an inadequate response to a single TNF-α antagonist, fostamatinib 100 mg bid, but not fostamatinib 100 mg bid for 4 weeks then 150 mg qd, achieved statistically significant improvements in ACR20 response rate at 24 weeks versus placebo. For the key secondary efficacy endpoints, ACR50 and ACR70, although statistical significance was obtained at Week 24 for Group A versus Group C, the number of patients achieving these response rates was low (compared with other RA therapies). Safety and tolerability findings were consistent with the profile observed in earlier studies6,7,8. The current results of fostamatinib in this phase III clinical study, along with those arising from the wider OSKIRA phase III clinical program, were not deemed sufficient and therefore the companies developing this particular SYK inhibitor decided not to study it further in RA at this time. It could be speculated that higher doses of fostamatinib may have been required to achieve levels of SYK inhibition that would achieve better levels of clinical efficacy, but the side effect/tolerability profile seen in phase II may have limited this.
Acknowledgment
We acknowledge the editorial services of Ellie Ling, PhD, from PAREXEL, which were funded by AstraZeneca.
Footnotes
-
Clinical study sponsored by AstraZeneca. M. Genovese has received research grants and consulting fees from AstraZeneca and Rigel. D. van der Heijde has received consulting fees from AbbVie, Amgen, AstraZeneca, Augurex, BMS, Celgene, Centocor, Chugai, Covagen, Daiichi, Eli Lilly and Co., GSK, Janssen, Merck, Novartis, Novo Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB, and Vertex, and serves as director at Imaging Rheumatology BV. E. Keystone has obtained research grants from Abbott, Amgen, AstraZeneca, Baylis Medical, Bristol-Myers Squibb, Roche, Janssen, Eli Lilly and Co., Novartis, Pfizer, Sanofi-Aventis, and UCB. He also has consulting agreements and/or advisory board membership with Abbott, AstraZeneca, Biotest, Bristol-Myers Squibb, Roche, Genentech Inc., Janssen Inc., Eli Lilly and Co., Merck, Nycomed, Pfizer, and UCB, and speaker honoraria agreements with Abbott, AstraZeneca, Bristol-Myers Squibb, Roche Inc., Janssen Inc., Pfizer, UCB, and Amgen. A. Spindler is a clinical study investigator for AstraZeneca. C. Benhamou has received research grants from Servier and Amgen and consulting fees from RottaPharm, and has held nonremunerative positions of influence for Novartis and Roche. A. Kavanaugh has received research grants from AbbVie, Amgen, AstraZeneca, Bristol-Myers Squibb, Roche, Janssen, Novartis, Pfizer, Sanofi-Aventis, and UCB. E. Fudman has received research grants from AstraZeneca, Astellas, Bristol-Myers Squibb, Genentech, Pfizer, and Sanofi-Aventis. E.L. Duffield is an employee of AstraZeneca and holds stocks/shares in AstraZeneca. K. Lampl and C. O’Brien are former employees of AstraZeneca and hold stocks/shares in AstraZeneca. J. Poiley has received research grants from AstraZeneca. M. Weinblatt has received research grants from Crescendo Bioscience, Bristol-Myers Squibb and UCB, has received consulting fees from AbbVie, Ablynx, Adheron Therapeutics, Amgen, Antares, AstraZeneca, Augurex, Bristol-Myers Squibb, Canfite, Crescendo Bioscience, Ensemble, Exagen, Five Prime, Genentech/Roche, Hutchison, Idera, Infinity, Janssen, Lycera, Lilly, Medimmune, Merck, Novo Nordisk, Pfizer, Regeneron, UCB and Vertex, and holds stocks/shares in Canfite, Ensemble and Lycera.
- Accepted for publication July 24, 2014.








{kind=link}
{kind=link}