

Abstract
Objective. To assess real-world safety, tolerability, and effectiveness of etanercept monotherapy, etanercept plus methotrexate (MTX), or etanercept plus other disease-modifying antirheumatic drugs (DMARD) in Japanese patients with active rheumatoid arthritis (RA) despite previous treatment with DMARD.
Methods. In this 24-week, all-cases postmarketing surveillance study, adverse events (AE) were coded using the Medical Dictionary for Regulatory Activities. Effectiveness was assessed every 4 weeks using the 28-joint Disease Activity Score and the European League Against Rheumatism response criteria.
Results. Of 13,861 patients (81% women) in the analysis, 3616, 2506, and 7739, respectively, were classified into etanercept monotherapy (ETN-mono), etanercept plus DMARD other than MTX (ETN + DMARD), and etanercept plus MTX (ETN + MTX) groups. Rates of AE and serious AE (SAE) in the ETN + MTX group were lower than in other groups. Risk of SAE or serious infections was not significantly increased with higher versus lower MTX doses at baseline or with concomitant use of salazosulfapyridine or bucillamine in ETN + DMARD versus ETN-mono groups. A greater likelihood of achieving clinical remission was seen with ETN + MTX versus ETN-mono (OR 1.36; 95% CI, 1.16–1.60; p < 0.001). Higher MTX dose at baseline was associated with a higher remission rate (> 8 mg vs 0 to ≤ 4 mg, OR 1.47, 95% CI 1.07–2.00, p = 0.016; 6 to ≤ 8 mg vs 0 to ≤ 4 mg, OR 1.27, 95% CI 1.01–1.60, p = 0.038).
Conclusion. Combination therapies with etanercept plus MTX or other DMARD were reasonably well tolerated, and ETN + MTX at higher doses was more effective than ETN-mono in Japanese patients with RA.
Rheumatoid arthritis (RA), a chronic inflammatory disease affecting joints and extraarticular tissues, is associated with pain, disability, and decreased life expectancy1,2. Among the newer treatments for RA, agents that inhibit tumor necrosis factor (TNF) have proven to be effective in controlling disease activity and reducing radiographic progression of the condition.
Etanercept is a recombinant, human, soluble, dimeric fusion protein that competitively binds to TNF and lymphotoxin-α and prevents its binding to endogenous receptors on the cell surface3. Randomized clinical trials have demonstrated the efficacy and safety of etanercept as monotherapy or combined with methotrexate (MTX), a disease-modifying antirheumatic drug (DMARD) that is now a standard treatment for RA4,5,6,7. Etanercept has shown efficacy superior to MTX in patients with RA7, and combining etanercept with MTX was superior to either agent alone6. In addition, the combination did not increase the risk for adverse events (AE) compared with monotherapy with either agent6.
A postmarketing surveillance (PMS) study in Japanese patients with RA was conducted by Wyeth (now integrated into Pfizer as of October 2009) under the request of the Japanese Pharmaceutical and Medical Device Agency (PMDA). This was a unique study in that all patients who were administered etanercept after its approval in Japan were enrolled in this PMS study for a 2-year survey period. Eventually, 13,894 patients were registered from 1334 medical sites. This study demonstrated the safety and effectiveness of etanercept in a real-world setting8.
We reported that the average dose of MTX in the PMS study was lower than that typically used in Western countries, and bucillamine (BUC), which has not been approved in Western countries, is one of the major DMARD in the PMS study8,9. Although our previous report of this PMS study showed that concomitant MTX seemed to result in better effectiveness and fewer safety problems8,10, the safety and effectiveness of concomitant use of DMARD other than MTX, such as BUC and sulfasalazine (SSZ), still have not been clarified. Further, the difference in safety and effectiveness between etanercept monotherapy and combination therapy has not yet been elucidated. In this analysis, we took advantage of the unique all-patients PMS study and the distinguishing characteristics of concomitant DMARD use in Japanese patients with RA to evaluate the safety and effectiveness of etanercept alone or in combination with MTX or with DMARD other than MTX in a large Japanese patient population.
MATERIALS AND METHODS
Patients
In the 2-year period from March 2005 to April 2007, 13,894 Japanese patients with RA participated in a 6-month PMS study of etanercept (ClinicalTrials.gov, NCT00503503). Candidates were deemed suitable for treatment with etanercept based on guidelines from the Japan College of Rheumatology11. Criteria for inclusion were active RA with at least 6 tender joints and at least 6 swollen joints and erythrocyte sedimentation rate ≥ 28 mm/h or C-reactive protein levels ≥ 2.0 mg/dl despite more than 3 months of previous treatment with DMARD8. In addition, participants were required to have leukocyte count ≥ 4000/μl, including peripheral blood lymphocyte count ≥ 1000/μl, and had to be negative for serum β-D-glucan (an indicator of immune activity). The dosage of etanercept in our study was determined by the discretion of the attending rheumatologists.
Assessments at study entry included chest radiographs, tuberculin tests, and medical history, including comorbid diseases. Characterization of RA was based on Steinbrocker radiographic stage and functional class12, duration of RA, and previous or current use of glucocorticoids or DMARD.
Patients were retrospectively classified into 3 treatment groups by concomitant use of DMARD at baseline: etanercept monotherapy (ETN-mono), etanercept plus DMARD other than MTX (ETN + DMARD), and etanercept plus MTX with or without DMARD other than MTX (ETN + MTX). Etanercept 10 or 25 mg was administered subcutaneously twice weekly (dosage determined by the physician). Patients were allowed to self-inject after training.
The protocol was reviewed and approved by the Japanese Ministry of Health, Labor, and Welfare. Data were collected electronically or on hard-copy case report forms, and representatives from Wyeth and Takeda visited study sites to collect additional data as required.
Assessments
Safety assessments were performed every 2 weeks and included recording of all AE occurring from the first etanercept dose to 30 days after the last dose. Safety data were coded with the matching terms from the Medical Dictionary for Regulatory Activities13. All AE, serious AE (SAE), serious infections (SI), and adverse drug reactions were defined based on International Conference on Harmonization guidelines14. Safety information was independently evaluated by the Japan College of Rheumatology PMS committee.
Treatment effectiveness was measured every 4 weeks using the 28-joint Disease Activity Score (DAS28)15 and the European League Against Rheumatism (EULAR) response criteria16. Missing data were processed using the last observation carried forward method, except for baseline values, which were not carried forward. The DAS28 scores were divided into 4 categories: remission (< 2.6), low disease activity (≥ 2.6 to ≤ 3.2), moderate disease activity (> 3.2 to ≤ 5.1), and high disease activity (> 5.1). EULAR responses were based on DAS28 results. A good response was defined as an improvement (i.e., a reduction in DAS28 score) of > 1.2 from baseline and DAS28 ≤ 3.2 at evaluation. A moderate response was defined as an improvement of 0.6 to 1.2 and DAS28 ≤ 5.1 at evaluation. Nonresponse was defined as an improvement of ≤ 0.6 or an improvement of 0.6 to 1.2 and DAS28 > 5.1 at evaluation. Treatment was deemed effective in patients with EULAR ratings of moderate or good response.
Patients < 17 years old were excluded from the study. For the effectiveness analyses, we also excluded patients who were treated for a nonapproved indication, whose treatment period was shorter than 2 weeks, whose DAS28 data either at baseline or 24 weeks were missing, or who had a DAS28 < 2.6 at baseline.
Statistical analysis
Baseline differences among the 3 treatment groups were assessed by 1-way ANOVA or chi-square tests. For each group, the change in DAS28 from baseline to each posttreatment assessment (every 4 weeks up to 24 weeks) was analyzed using the Jonckheere Terpstra test, and the change in response rates was assessed by Cochran-Armitage tests. Differences between groups on DAS28 at baseline and each posttreatment assessment point (every 4 weeks) were analyzed by 1-way ANOVA and Dunnett multiple comparison tests, and differences in EULAR responses rate at each posttreatment assessment point were analyzed by chi-square tests. Differences between groups on DAS28 improvement from baseline to Week 24 were analyzed by 1-way analysis of covariance, with combination treatment as factors and the baseline value as a covariate. The incidence of most common AE and SAE among the 3 subgroups were assessed by chi-square test or Fisher’s exact test (when n < 5) and used the Bonferroni correction to adjust the result of multiple testing.
Cox proportional hazard models were used to estimate the influence of combination with DMARD (concomitant use of MTX or DMARD other than MTX) on the occurrence of SAE and SI, after adjusting for the following major confounders: age, sex, history of infectious disease, history of tuberculosis, previous use of infliximab, Steinbrocker class, and presence of combined risk factors. In patients who had 2 or more SAE, only the first SAE was counted for Cox proportional hazard models.
The combined risk factors included 3 risk factors that are indicated by our previous report17: comorbidities, concomitant glucocorticoid use, and disease duration > 15 years. Association of the combined effect and numbers of risk factors were further explored by the Wald test.
We also used multiple logistic regression models to estimate the effect of concomitant DMARD use (concomitant use of MTX or DMARD other than MTX) on the likelihood of achieving remission and good response after adjusting for the following major confounders: age, sex, baseline disease activities, previous use of infliximab, presence of any comorbidities, Steinbrocker functional class, and duration of RA. Treatment effectiveness was assessed in treated patients with DAS28 evaluated at baseline and at 24 weeks. Patients were excluded from these models based on missing DAS28 data either at baseline or at 24 weeks, DAS28 < 2.6 at baseline, or missing data for adjustment factors (e.g., age, sex, disease duration).
Further, we estimated the effect of MTX dosage on safety and effectiveness. The HR or OR and the 95% CI for each factor after adjustment for major confounders were estimated. All statistical analyses were performed using SAS software version 9.2 (SAS Institute Inc.). Statistical significance was defined by 2-sided p values < 0.05.
RESULTS
Patients
A total of 13,894 patients were treated with etanercept alone or in combination with DMARD. Overall, 13,861 patients who were older than 17 years of age at baseline in the treated population were evaluated for safety and tolerability in the 3 treatment groups: ETN-mono (n = 3616, 26.1%), ETN + DMARD (n = 2506, 18.1%), and ETN + MTX (n = 7739, 55.8%). The numbers of patients with concomitant use of 1, 2, 3, and 4 or more DMARD including MTX were 7801 (56.3% of total patients), 2165 (15.6%), 247 (1.8%), and 32 (0.2%), respectively. The percentage of patients who received SSZ or BUC in the ETN + MTX groups (18.8%) was significantly lower than in the ETN + DMARD groups (61.7%). Patient numbers with SSZ and/or BUC are described in detail in Figure 1. Of the 13,861 patients, 7325 patients with available data were evaluated for effectiveness (Figure 1).
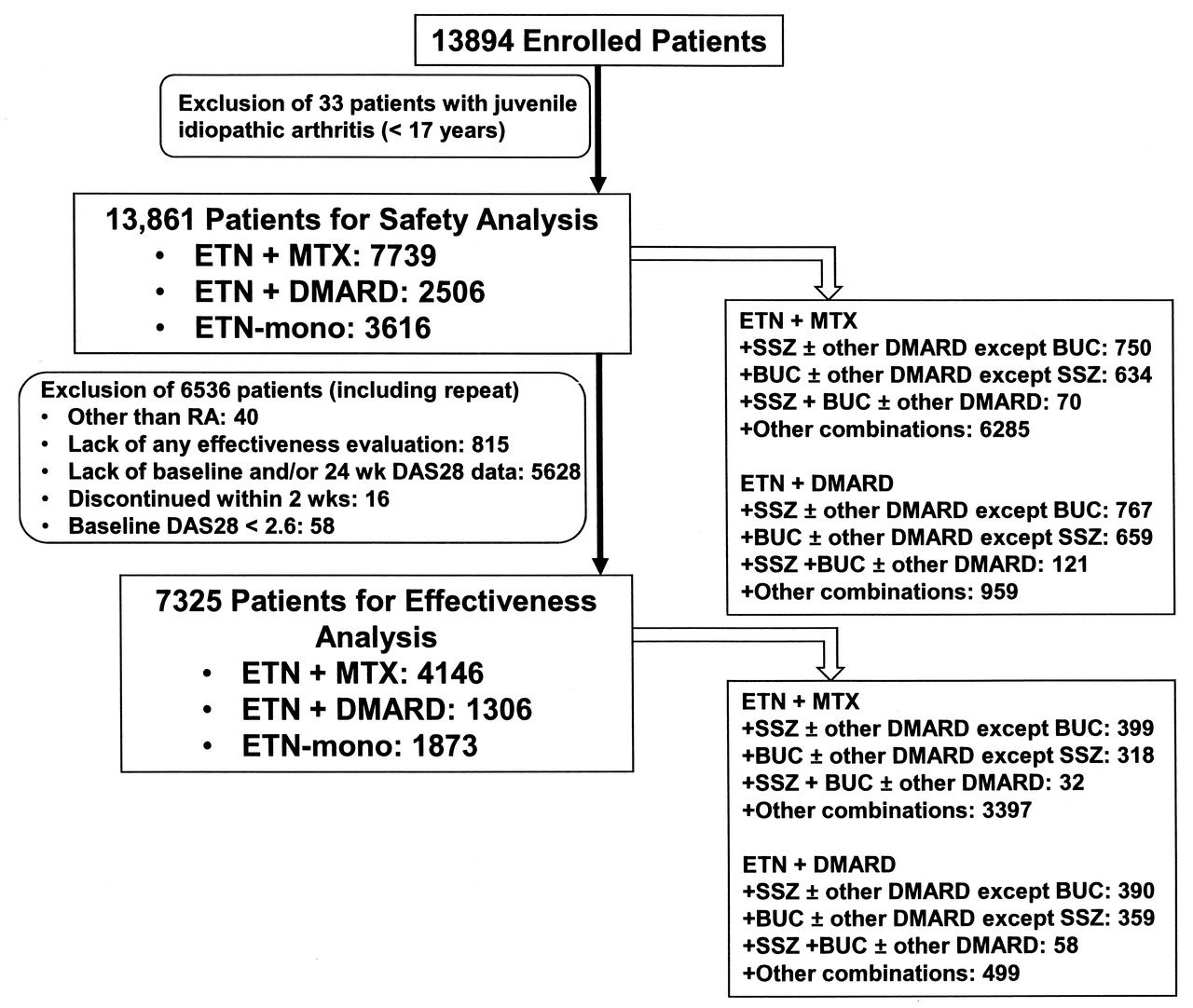
Flow chart of the evaluated patients.
Among the 3 groups, most of the demographic and baseline disease characteristics were significantly different, such as age, disease activity, disease duration, comorbidities, and concomitant corticosteroid, etc. The mean age, disease activity (DAS28), and the percentage of patients with comorbidities and history of selected diseases were significantly lower in the ETN + MTX group than in the other groups. The previous use of infliximab was significantly higher in the ETN + MTX group than those in the other groups. Most of the demographic and baseline disease characteristics were similar between the ETN + DMARD and the ETN-mono groups (Table 1).
Demographic and baseline disease characteristics of patients included in the safety analysis.*
Safety
Compared with the ETN-mono and the ETN + DMARD groups, the ETN + MTX group showed significantly lower incidence rates for total AE, total SAE, the 2 most frequently observed AE, and the 4 most frequently observed SAE (Table 2). Further, both of the incidences of AE and SAE were significantly lower with MTX than with no MTX (both p < 0.001). Within the ETN + MTX group, for AE and SAE, there was no evidence of significant correlation between incidence and increasing MTX dosage (both trend p > 0.05; Table 3).
Incidence of most common AE and SAE among the 3 subgroups.
Incidence of AE and SAE among the MTX dose subgroups.
We initially implemented Cox proportional hazard regression model using all patients for safety analysis. Compared with the ETN-mono group, the risks of SAE and SI were significantly lower in the ETN + MTX group (HR, 0.59 and 0.61; 95% CI, 0.50–0.70 and 0.47–0.79, respectively; all p < 0.001) but not in the ETN + DMARD group (data not shown). These findings are compatible with our previous report10. Considering significant difference in the demographic and baseline disease characteristics among the 3 groups, we postulated that analyzing all patients in 1 statistical model for safety was not satisfactory and conducted Cox proportional model analysis for safety for each group.
Risk factors for the development of SAE and SI within the ETN + MTX group are shown in the model for baseline characteristics in Table 4. These results were similar to those previously reported10. The results in the model for MTX dosage in Table 4 showed that higher MTX dose at baseline did not significantly increase the risk of SAE and SI.
Risk factors for SAE and serious infections in the ETN + MTX group.
Risk factors for the development of SAE and SI within the ETN + DMARD group are shown in Table 5. The results were somewhat different from those of the ETN + MTX group.
Risk factors for SAE and serious infections in the ETN + DMARD group. Multivariate analysis was performed on 2257 cases.
Within the ETN + DMARD group and the ETN-mono group, proportional hazards models indicated that both concomitant use of SSZ with or without other DMARD (ETN + SSZ) and concomitant use of BUC with or without other DMARD except SSZ (ETN + BUC) was not significantly associated with the risk for SAE and SI when comparing with the ETN-mono group (data not shown).
Effectiveness
Similar to the results previously reported for all subjects10, for all treatment groups, the mean DAS28 scores, the DAS remission rate, and the EULAR good response rate showed a trend of significant improvement throughout the observation period. The EULAR good response rate at 24 weeks of the ETN + MTX group was significantly greater than those of the ETN-mono group or the ETN + DMARD group (29.8%, p = 0.003 at Week 24); further, there were no significant differences in improvement between the ETN-mono and the ETN + DMARD groups at 24 weeks (data not shown).
The DAS28 remission rate (< 2.6) was increased with higher dosages of MTX (Figure 2A). The remission rate for patients who received MTX > 10 mg weekly was 29.6%, about twice as high as that of patients who received no MTX. The EULAR good response rate increased with higher dosages of MTX (Figure 2B), from 27.2% at 0 mg weekly to 46.8% at > 10 mg weekly.

Effectiveness for the first 24 weeks in patients receiving etanercept monotherapy or etanercept plus MTX by categories of MTX dosage at baseline as indicated by (A) disease activity status and (B) EULAR response criteria. The trend on remission rate and response rate was significant (p < 0.001, Cochran-Armitage test). EULAR: European League Against Rheumatism; MTX: methotrexate.
Next, we focused on the effects of concomitant DMARD and clinical response to treatment with ETN. Concomitant use of MTX was associated with a greater likelihood of achieving significant clinical remission, whereas concomitant use of DMARD other than MTX, including SSZ and BUC, did not show statistically significant differences (Table 6). With regard to other variables, our results were similar to those previously reported10. Further, results of the multiple logistic regression model within the ETN + MTX group showed that higher MTX dose at baseline was associated with a higher remission rate than lower MTX doses at baseline as well as higher rate of good response.
Effect of concomitant DMARD at baseline on clinical response during treatment with etanercept. “No” refers to patients without concomitant use of any DMARD.
DISCUSSION
This PMS study was one of the largest surveillance studies of biologic use in rheumatology, with nearly 14,000 patient registrations. Mandatory registration for all patients with RA receiving etanercept regimens in Japan covered almost all patients treated with etanercept during the 2-year study duration. Almost three-quarters of the 13,861 patients were treated with etanercept plus at least 1 DMARD and over half of patients were treated with etanercept plus MTX. Therefore, this PMS study provided a unique and valuable opportunity to evaluate the real-world safety and effectiveness data for etanercept with and without DMARD in a large number of patients in Japan. DMARD, such as MTX, BUC, and SSZ, are often used as treatment for patients in Japan with RA. When patients are not achieving adequate response or cannot continue these DMARD because of toxicity, TNF inhibitors are usually prescribed for further treatment, as add-ons or substitutes. Therefore, it is important to evaluate the safety and effectiveness of etanercept with or without these DMARD.
As described, among the patients in this PMS study, occurrence rates of AE, SAE, and SI were comparable to those seen in clinical trials and registries17. In this PMS study of Japanese patients with active RA despite previous treatment with DMARD, treatment with etanercept in combination with MTX was at least as safe and well tolerated as etanercept monotherapy or etanercept plus DMARD other than MTX, as assessed by the incidences of overall and 5 most common AE and SAE. We should carefully compare these incidence rates because of the significant difference in demographic and baseline disease characteristics of patients such as age, disease activity, disease duration, comorbidities, and concomitant corticosteroid use among the 3 groups. Results of the Cox proportional hazard model indicated that treatment with etanercept plus different doses of MTX did not alter risk for SAE and SI in the ETN + MTX group. Risk factors for SAE and SI found in the ETN + MTX group (male sex, older age, Steinbrocker class 4, concomitant disease, concomitant glucocorticoid use, and long disease duration for both SAE and SI; concomitant nonserious infections for SI only) were also reported as risk factors for etanercept use in the RADIUS registry17.
Results of the Cox proportional hazard model also indicated that there were differences in the risk factors for SAE and SI between the ETN + MTX groups and the ETN + DMARD group. The results show no significant difference in the ETN + DMARD group for SAE and SI in a higher Steinbrocker class and for SI in the group with > 1 selected-risk factor, but statistically significant results in the ETN + MTX group for this specific aspect. Although there are several possible reasons, such as the differences in baseline characteristics between the ETN + DMARD group and the ETN + MTX group, the sample size is not large enough to show significant difference. These results suggested that it is better to carefully observe the SAE and/or SI when ETN + MTX was used to treat patients with higher Steinbrocker class and/or with > 1 selected-risk factor.
Etanercept alone or in combination with other DMARD was also effective for improving RA symptoms, as assessed by DAS28 measurement of disease severity and EULAR response categories from Week 4, and this improvement had not yet plateaued at Week 24 (data not shown). The EULAR good response rate was significantly higher in the ETN + MTX group compared with the ETN-mono group or the ETN + DMARD group. Superiority of etanercept plus MTX compared with etanercept monotherapy in our study is consistent with the results from previously reported clinical trials4,6. Because the background characteristics of the patients, which may affect effectiveness of ETN, were significantly different among the 3 groups, we adjusted for age, sex, Steinbrocker class, disease duration, previous use of infliximab, and baseline DAS28 and showed favorable effect of MTX to achieve better clinical response (Table 6).
Numerous clinical trials have demonstrated that aggressive combination therapy early in the course of RA may result in higher rates of clinical remission and less radiographic progression than monotherapy, conventional sequential therapy, or step-up therapy4,18,19,20,21,22,23. In our study, the remission rate was significantly greater with etanercept plus MTX than with etanercept alone.
Further, a positive correlation was suggested between MTX dosage and a favorable clinical outcome. Results of the Cox proportional hazards model indicated that patients receiving higher dosages of MTX had a higher probability of achieving clinical remission. Similar results were also reported in another study24. In addition, higher dosages of MTX did not alter the risk for SAE or SI, even though the dosage was > 8 mg weekly. These findings are especially noteworthy because MTX dosages are typically lower in Japan than in Western countries, where dosages of 15 to 20 mg weekly are often used9; relatively few of the patients in this Japanese study received MTX > 8 mg weekly. Yamanaka, et al reported that there was a positive correlation between MTX dosage and the frequency of adverse reactions with MTX in the IORRA cohort9. It was previously reported from the Japanese REAL cohort that the use of MTX > 8 mg weekly was a risk factor for SI25. In February 2011, the higher dosage of up to 16 mg weekly was approved by PMDA in Japan. Further investigations are expected in exploring safety and effectiveness of MTX at this higher dose in the Japanese population.
Although the DAS28 remission rate observed in the ETN + MTX group in our study (21%; data not shown) was lower than that observed in other clinical studies (35%)6, the favorable risk/benefit ratio observed with combination etanercept plus MTX suggests that this approach is warranted in many patients with RA who can tolerate MTX. Indeed, aggressive combination regimens offer a high level of effectiveness and tolerability, which will make remission a realistic goal in many patients with RA26.
Our study provided unique results in the clinical use of lower dosages of MTX with etanercept. Because MTX is usually prescribed at more than 8 mg weekly in many countries but not in Japan, results for the lower dosage of MTX have not been well documented. Therefore, comparing safety and effectiveness between ≤ 8 mg weekly and > 8 mg weekly MTX plus etanercept is applicable to real-world MTX use of etanercept in Japan. This was consistent with the result of the JESMR study27. In that study, ≤ 8 mg weekly MTX plus etanercept was superior to etanercept monotherapy for inadequate responders to MTX.
Our results indicated that concomitant use of SSZ or BUC did not alter the risk for SAE and SI (Cox proportional hazard model; data not shown), and concomitant use of SSZ or BUC did not affect the remission rates (multiple logistic regression model). Combe, et al reported that concomitant use of SSZ with etanercept did not alter the incidence of AE, but the incidence of infection was significantly lower with combination therapy28. Regarding efficacy, etanercept plus SSZ was significantly more sustainable and efficacious for DAS improvement after 68 weeks of treatment compared with etanercept monotherapy28. In the Combe, et al study, duration of treatment was 2 years and the dosages of SSZ were 2, 2.5, and 3 g daily, which are higher than the typical dosage of SSZ (1 g daily) in Japan28,29,30,31. Shorter treatment duration and/or lower dosage of SSZ in our PMS study may explain this discrepancy. Regarding BUC, a DMARD approved only in Japan and Korea31,32, evidence supporting its concomitant use with etanercept has not been established. Only the safety and efficacy of concomitant use of etanercept plus SSZ plus BUC has been reported33. In that study, the efficacy of concomitant use of etanercept plus SSZ or BUC was higher than that of etanercept monotherapy and comparable to that of etanercept plus MTX. Further studies are needed to evaluate the safety and effectiveness of concomitant use of these DMARD with etanercept.
As in our previous report10, the main limitation of our current study was the absence of a comparator group and presence of indication bias for concomitant DMARD. PMS studies do not always include a comparator group, which makes it difficult to distinguish outcomes relating to ETN + MTX treatment from those caused by confounding factors. Another important limitation was that about 50% of patients were excluded from effectiveness analysis because of a lack of DAS28 data at baseline and/or Week 24. This exclusion makes it difficult to generalize the outcomes of effectiveness. The short duration of the 6-month PMS study was also an important limitation. Although most of the previously reported AE were detected and a general safety profile of etanercept was obtained, AE that require a longer followup period, such as malignancy, can hardly be evaluated. For this reason, we have conducted another longterm PMS study to evaluate AE, including malignancy. The results of this longterm study will be reported soon.
We used the MTX weekly dose at baseline to evaluate the influence on effectiveness without considering changes of dosage during the treatment period. The analysis with mean dosage of MTX may be necessary. In addition, radiographic findings were not included and the observational period was relatively short (6 months). There are also some limitations on statistical methodology. For Table 1, we have not considered the issue of multiple testing, so it is possible that the significant difference in some demographic and baseline disease characteristics will disappear when the correction is used. Despite these limitations, this all-cases, 2-year PMS study demonstrates the real-world safety and effectiveness of etanercept-DMARD combination therapy with data from about 14,000 patients.
Both etanercept monotherapy and etanercept in combination with DMARD are effective in improving RA disease control and are reasonably well tolerated. However, the best responses were observed in patients who received etanercept in combination with MTX, especially in those who received doses of MTX higher than those typically used in Japan. Our findings support the use of this treatment approach to improve RA control, establishing remission as a realistic goal for many patients.
Acknowledgment
We thank all investigators for contributions to this study and acknowledge the contributions of the late Professor Kazuhiko Inoue, MD, PhD, who participated in this work. This report was prepared with the assistance of Joanne Foehl (Pfizer Inc., Global Medical Affairs) and Complete Healthcare Communications Inc., Chadds Ford, Pennsylvania, USA.
Footnotes
-
Supported by Wyeth; clinical fees were shared by Wyeth K.K. and Takeda Pharmaceutical Company Ltd. T. Koike, M. Harigai, S. Inokuma, N. Ishiguro, J. Ryu, T. Takeuchi, Y. Tanaka, and H. Yamanaka are all members of the Etanercept Postmarketing Surveillance Committee of the Japan College of Rheumatology. Financial relationships of authors with all manufacturers of products used in the management of rheumatoid arthritis are as follows. T. Koike: consulting fee from UCB Japan Co. Ltd.; on the speakers’ bureau of Abbott Japan Co. Ltd., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Takeda Pharmaceutical Co. Ltd., Pfizer Japan Inc. M. Harigai: research grant to the affiliated institution from AbbVie G.K., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd.; consulting fee from AbbVie G.K., Chugai Pharmaceutical Co. Ltd., Mitsubishi Tanabe Pharma Corp.; on the speakers’ bureau of AbbVie G.K., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd. S. Inokuma: none. N. Ishiguro: research grant to the affiliated institution from Abbott Japan Co. Ltd., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi-Sankyo Pharmaceutical Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd.; on the speakers’ bureau of Abbott Japan Co. Ltd., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi-Sankyo Pharmaceutical Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd. J. Ryu: none. T. Takeuchi: research grant to the affiliated institution from AbbVie G.K., Abbott Japan Co. Ltd., Asahi Kasei Pharma Corp., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Eisai Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Sanofi K.K., Santen Pharmaceutical Co. Ltd., Taisho Toyama Pharmaceutical Co. Ltd., Takeda Pharmaceutical Co. Ltd., Teijin Pharma Ltd.; consulting fee from AbbVie G.K., Asahi Kasei Pharma Corp., AstraZeneca K.K., Daiichi Sankyo Co. Ltd., Eli-Lilly Japan K.K., Mitsubishi Tanabe Pharma Corp., Novartis Pharma K.K.; on the speakers’ bureau of Abbott Japan Co. Ltd., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Eisai Co. Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd. Y. Tanaka: research grant to the affiliated institution from AbbVie G.K., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Mitsubishi Tanabe Pharma Corp., MSD; on the speakers’ bureau of AbbVie G.K., Abbott Japan Co. Ltd., Asahi Kasei Pharma Corp., Astellas Pharma Inc., AstraZeneca K.K., Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Eisai Co. Ltd., Eli Lilly Japan K.K., GlaxoSmithKline K.K., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corp., MSD, Pfizer Japan Inc., Quintiles Inc., Takeda Pharmaceutical Co. Ltd. H. Yamanaka: research grant to the affiliated institution from AbbVie G.K., Abbott Japan Co. Ltd., Asahi Kasei Pharma Corp., Astellas Pharma Inc., Bristol-Myers Squibb, Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Eisai Co. Ltd., GlaxoSmithKline K.K., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corp., MSD, Nippon Kayaku Co. Ltd., Pfizer Japan Inc., Santen Pharmaceutical Co. Ltd., Taisho Toyama Pharmaceutical Co. Ltd., Takeda Pharmaceutical Co. Ltd., Teijin Pharma Ltd.; on the speakers’ bureau of Abbott Japan Co. Ltd., Chugai Pharmaceutical Co. Ltd., Daiichi Sankyo Co. Ltd., Mitsubishi Tanabe Pharma Corp., Pfizer Japan Inc., Takeda Pharmaceutical Co. Ltd., Teijin Pharma Ltd. T. Hirose, T. Yoshinaga, and M. Suzukawa are fulltime employees of Pfizer Japan Inc.
- Accepted for publication June 6, 2013.








{kind=link}
{kind=link}