

Abstract
Tumor necrosis factor (TNF) antagonists are drugs developed to block endogenous TNF, an essential proinflammatory molecule with a central role in the pathogenesis of rheumatoid arthritis (RA). Although extensive studies have been performed concerning the mode of action of TNF-blocking agents, there are still many unresolved questions and potential differences between different TNF-blocking drugs. One unresolved issue is to what extent apoptosis is affected by TNF blockade in RA. We provide an overview of studies that have investigated the proapoptotic effect of different anti-TNF drugs in RA, searching for a unified interpretation of somewhat contradictory data.
Tumor necrosis factor (TNF) was discovered as a potential key modulator in rheumatoid arthritis (RA) more than a decade ago1. Subsequent studies confirmed that TNF is present in both synovial tissue2 and synovial fluid as well as serum3 of patients with RA, and identified synovial macrophages as the principal source of TNF4. Animal studies offered a deeper understanding of the implications of TNF in the pathogenesis of RA. TNF transgenic mice develop spontaneous RA-like disease with synovial inflammation and joint destruction5. Moreover, administration of TNF aggravates disease progression in mouse strains susceptible to arthritis6. But perhaps the most convincing evidence regarding the key role of TNF in RA pathogenesis is the high clinical efficacy of different TNF-blocking therapies in patients with RA7,8. Several TNF antagonists are currently available for clinical use: etanercept (a recombinant TNF receptor-Fc fusion protein), infliximab (a chimeric monoclonal antibody), adalimumab (a fully human anti-TNF antibody)7, certolizumab pegol (a pegylated humanized Fab fragment)9, and golimumab (a human anti-TNF antibody)10,11. All these drugs were originally designed to block TNF, but differences in their mechanisms of action might result from their ability to bind different types of TNF molecules and from their different structures. Infliximab and adalimumab bind with high affinity to both soluble and membrane TNF, and are able in vitro to induce both antibody-dependent cell-mediated cytotoxicity (ADCC)12,13 and complement-dependent cytotoxicity (CDC). Etanercept binds primarily soluble TNF and to a lesser extent cell-surface TNF14. It has been suggested that etanercept does not mediate either ADCC or CDC15,16 despite presence of an Fc portion in the molecule. However, another report demonstrated that etanercept can induce both ADCC and CDC in a stably transfected cell line expressing TNF-α17. Golimumab is an IgG1 antibody therefore able to induce both ADCC and CDC18, while certolizumab is the only pegylated Fab anti-TNF lacking the Fc portion and therefore is unable to mediate either of these effects17. Direct comparison of TNF binding affinities demonstrated that infliximab, adalimumab, etanercept, and certolizumab pegol bind to transmembrane TNF-α on transmembrane TNF-α-transfected cells17,19,20 with similar affinities that were weaker than for soluble TNF-α21. The bioactivity of the IgG1 molecules golimumab, infliximab, and adalimumab in neutralizing soluble human TNF is inferior to that of certolizumab and etanercept. Certolizumab pegol is the most potent at inhibiting lipopolysaccharide (LPS)-driven production of interleukin 1ß (IL-1ß) by monocytes, followed by golimumab, adalimumab, and infliximab, while etanercept has only a partial effect22. Infliximab and adalimumab have similar efficacy profiles and except for RA are used in more inflammatory diseases such as psoriatic arthritis (PsA), psoriasis, ankylosing spondylitis (AS), and Crohn’s disease. Etanercept lacks efficacy in granulomatous diseases such as Crohn’s and granulomatosis with polyangiitis (Wegener’s) and may be less efficacious in psoriasis8. Golimumab is approved for treatment of RA, PsA, and AS23. Certolizumab pegol is approved for treatment of RA and Crohn’s disease24.
RA and Apoptosis
Defective apoptosis of resident cells contributes to excessive synovial cell infiltration and perpetuation of chronic inflammation in RA25. Histological studies26,27 demonstrated low levels of apoptosis in the RA synovial tissue, between 1% and 3% of synovial cells, despite the presence of both cell death receptors (Fas and TNFR) and cell death ligands (Fas-ligand and TNF)2,28,29 at the site of the inflamed synovium. It was originally suggested that concomitant occurrence of soluble forms of the receptors and/or their ligands might act as endogenous inhibitors30,31, while another study claimed deficient expression of functional FasL on synovial lymphocytes32. It is thought today that resistance to cell death receptor-mediated apoptosis in the inflamed synovium is largely due to high expression of inhibitory molecules such as Fas-associated death domain-like IL-1ß-converting enzyme inhibitory protein (FLIP)33,34, nuclear factor-κB (NF-κB) transcription factor35,36, and sentrin37. Concomitant inhibition of the mitochondrial pathway due to synovial expression of the antiapoptotic but not proapoptotic Bcl-2 family members38,39, as well as presence of dominant negative p53 mutants40 despite high p53 synovial expression41, contribute further to the apoptosis-resistant phenotype of the RA synovium. Recently, synovial expression of inhibitors of apoptosis proteins, blocking both cell death-mediated and mitochondrial pathways at the common level of caspase 3, has been reported in RA and was correlated with the levels of synovial apoptosis42.
RA and TNF-mediated Apoptosis
TNF is a key agent of innate immunity and an important modulator of inflammation. It belongs to the TNF superfamily and consists of a 26-kDa protein expressed on the cell surface or present in a soluble form following cleavage by a protease called TNF-α-converting enzyme43. Both membrane-bound and soluble forms are biologically active. The effects of TNF are mediated by 2 structurally distinct receptors: type I (TNF-RI: p60 or p55) and type II (TNF-RII: p80 or p75). Under different conditions, such as inflammation, these receptors are shed from cell surfaces and released into the circulation. Both TNF-RI and TNF-RII have high affinity for TNF but the rate of dissociation is higher for TNF-RII44. TNF ligation induces trimerization of cell-surface receptors, followed by intracellular signaling. There are 2 main intracellular pathways activated by TNF, resulting in activation of either the transcription factor NF-κB following expression of survival genes, or activation of caspases following apoptosis45. These 2 pathways are closely linked as far as inhibition of constitutive NF-κB activation, in the absence of an additional cell death-inducing signal, may by itself result in cell apoptosis46. Although both receptors can transduce the signaling pathways for apoptosis and NF-κB activation, TNF-RI is responsible for these signals in most cases47.
Apoptosis can be modulated not only by surface TNF receptors but also through reverse signaling upon ligation of transmembrane TNF by its counter-receptor/antibodies48,49. One potential intracellular pathway activated through this mechanism is dependent on p5348 and further induction of proapoptotic Bcl-2 family members48,50. Recently, it has been shown that reverse signaling also leads to NF-κB suppression and apoptosis induction apparently through a caspase-independent mechanism51. However, NF-κB blocking in RA synovial fluid cells downmodulates FLIP-like genes52, suggesting that reverse signaling might promote even a caspase-dependent apoptosis. Other effects mediated through reverse signaling inhibition of LPS-driven cytokine production by monocytes53 and induction of neutrophil necrosis54.
TNF Antagonists and Synovial Apoptosis
TNF antagonists decrease the macroscopic inflammatory joint score as well as microscopic inflammation, in terms of reduction of the number of synovial inflammatory cells. High-dose infliximab decreases both synovial lymphocyte and macrophage numbers55,56, while doses of 3 mg/kg appear to decrease only macrophage numbers57,58. Etanercept decreases synovial macrophages but not lymphocyte numbers58. Adalimumab decreases macroscopic inflammation in the rheumatoid joint without significant changes in synovial cellularity according to classical histological score59, although studies regarding specific inflammatory cell subsets are not available. We currently lack histological studies on the effect of the other 2 TNF antagonists (certolizumab and golimumab) that were introduced into daily clinic use relatively recently. The observed reduction in the number of immune cells at the site of inflammation following use of TNF antagonists might be due to decreased local cell recruitment and/or to increased clearance of resident cells, through induction of cell death or increase of cellular efflux. Several studies have demonstrated that infliximab, adalimumab, and etanercept are all able to decrease leukocyte trafficking to the joint. Infliximab, for example, reduces granulocyte recruitment and might interfere with monocyte trafficking through downregulation of synovial monocyte chemotactic protein-1 and expression of cell adhesion molecules55,56. Both infliximab and etanercept increase peripheral CXCR3-positive T cells, an indirect proof for decrease of recruitment of T cells to the site of inflammation60. Adalimumab decreases the influx of leukocytes into the joint without impairing neutrophil chemotaxis ex vivo61. Decreased recruitment of peripheral blood cells to the inflammation site should theoretically result in an increase in peripheral blood cell counts. However, 1 week of infliximab treatment induces a decrease in monocyte and lymphocyte counts62,63, while 2 weeks of etanercept treatment does not significantly alter peripheral blood counts64, suggesting that other mechanisms than decreased cellular trafficking should contribute to the observed decrease in cellularity. One possibility is an increase in the cellular efflux, but this hypothesis is still highly speculative, with only 1 study suggesting an increase in lymph vessel formation following treatment with infliximab65. The second possibility is increased apoptosis. One study exploring induction of apoptosis by infliximab in patients with RA failed to demonstrate any changes in synovial apoptosis as late as 28 days after the first infusion57. A followup study also failed to identify any increase in synovial apoptosis as early as 1 hour after the first infusion66. However, 8 weeks of treatment with both infliximab and etanercept resulted in an increased number of synovial tissue apoptotic cells58. One possible explanation for the reported differences might be the timing of the followup. Even though induction of apoptosis is a rapid event, altering the status of an antiapoptotic environment such as the rheumatoid synovium might require complex changes. Effects of such complex changes might thus become evident as a change in apoptotic cells only at later timepoints, including the 8 weeks of treatment (with 2 weeks after the last infliximab infusion), as in our study58. This hypothesis is strengthened by a study in Crohn’s disease, demonstrating an increased level of apoptosis following 10 weeks of infliximab therapy compared to baseline67. The low numbers of patients in the above-noted studies (n = 1257, n = 566, and n = 2158) limit the possibility of a definite conclusion and require further investigation in larger cohorts.
TNF Antagonists and Apoptosis Outside the Synovial Membrane
In vitro studies have been more successful in finding some consensus on the apoptosis-inducing capacity of TNF antagonists. We were the first to report that both infliximab and etanercept induced in vitro apoptosis of monocytes, but not lymphocytes, derived from RA synovial fluid and to a lesser extent apoptosis of monocytes derived from RA peripheral blood following 24-hour incubation58. This observation was subsequently partly confirmed for infliximab, which induced apoptosis of monocytes derived from peripheral blood of subjects with RA but not healthy individuals following 48-hour incubation51. In a more recent report, both infliximab and adalimumab and to a lesser extent etanercept had the ability to induce apoptosis of activated peripheral blood lymphocytes and monocytes, while certolizumab pegol exhibited no such effects17. As well, infliximab and adalimumab, but not etanercept, induced apoptosis of a human monocytic cell line in vitro and in vivo after transfer in a chimeric human-mouse model68. Crosslinking of the etanercept bound to membrane TNF with the help of anti-human anti-IgG resulted in increased apoptosis and suggested a role for multimer formation in TNF antagonist-mediated apotosis48. However, in a recent report etanercept and to a lesser extent infliximab and adalimumab were able to induce apoptosis in synovial membrane-derived fibroblast cells in the presence of autologous peripheral blood mononuclear cells69. Taken together, the in vitro data support a direct effect of TNF antagonists on apoptosis rather than an indirect in vivo effect as a consequence of changes in the inflammatory synovial milieu. While in vitro testing demonstrated that TNF antagonists can induce apoptosis outside the synovial membrane, with some differences potentially related to the agent used, the cell type, and the cell activation state, in vivo studies have been less successful, with contradictory results. One study that investigated the level of apoptosis induced by infliximab in peripheral blood of RA patients 1 and 24 hours after drug administration and immediately after blood sampling failed to demonstrate any changes66. Another study suggested that TNF antagonists increase the level of peripheral blood nucleosomes, an indirect measure of apoptosis, but inclusion of patients with other diagnoses than RA limited interpretation of the study70. One potential limitation of the in vivo approach is the large array of confounding factors that might influence apoptosis levels in the peripheral blood, thus preventing detection of a real difference.
Interestingly, apoptosis outside the synovial membrane has also been suggested to play an important role in determining the side effect profile of distinct TNF antagonists. It is proposed, for example, that antibodies against TNF are able to induce apoptosis of T cells and to mediate CDC and ADCC, allowing expansion of immunosuppressive regulatory T cells with a potential reduction of interferon-γ responses that might lead to increased susceptibility to tuberculosis reactivation as compared to soluble receptors (for a review, see Harris and Keane71).
TNF Antagonists and Apoptosis — a Drug-specific Effect?
With the background of different effects observed with different conditions and with different TNF antagonists it has been suggested that induction of apoptosis might be a drug-specific rather than a class-specific mechanism of action. This hypothesis originated in the original investigation on apoptosis induction in Crohn’s disease, where infliximab induced apoptosis of activated lamina propria T lymphocytes72, activated peripheral blood lymphocytes73, and peripheral blood monocytes74, while no such effects were observed for etanercept75,76. However, an alternative explanation for the lack of effect of etanercept on Crohn’s-derived mononuclear cells is a cell-specific effect, where etanercept that binds transmembrane TNF with a lower affinity than infliximab and adalimumab19 might only be able to induce apoptosis in cells with high transmembrane TNF (tmTNF) expression, such as synovial macrophages, but not in cells with lower levels of tmTNF such as intestinal macrophages or monocytic cell lines77. In accord with previously presented data, etanercept and infliximab do not have an effect on lymphocyte apoptosis in RA58, while in Crohn’s disease infliximab but not etanercept increases apoptosis of activated lymphocytes72,75. A recent report further demonstrated that infliximab, adalimumab, and certolizumab, but not etanercept, induce apoptosis in CD4-positive intestinal lymphocytes when cocultured with CD14-positive peripheral autologous cells from patients with inflammatory bowel diseases78. The same reasoning might explain different effects of the same drug (in this case infliximab) when tested on different types of cells, with induction of apoptosis in Crohn’s but not RA lymphocytes. Intestinal lymphocytes in Crohn’s are highly activated and able to produce high levels of cytokines79. We and others2,80,81 have demonstrated that lymphocytes derived from both synovial tissue and synovial fluid express low levels of cytokines and show signs of anergy. Repeated in vitro treatment with TNF, a setting that mimics the chronic exposure to TNF in the RA synovial environment, suppresses T cell activity82. In support of the cell-specific hypothesis is the finding that TNF antagonists exhibit opposite effects in distinct cells derived from the same disease, with induction of apoptosis in activated mononuclear cells and prevention of apoptosis in colonic epithelial cells83,84,85 of patients with Crohn’s disease. The pathogenesis of Crohn’s disease consists of high apoptosis of epithelial cells and infiltration with enhanced survival of mononuclear cells, and we propose that TNF antagonists can reverse these 2 aberrant mechanisms. This implies that TNF antagonists differentially regulate distinct cell types with different status of activation and different sensitivity to apoptosis83. The potential effect of TNF antagonists on apoptosis has also been studied in other inflammatory diseases such as psoriasis and PsA86,87 and spondyloarthropathies88, as well as noninflammatory conditions such as diabetes-associated skin ulcerations89 and ventilator-induced lung injury90, but these findings are beyond the interest of this report.
TNF antagonists induce apoptosis in certain cellular systems and conditions through reverse signaling (Figure 1). The effect is dependent on the activation status of the cells and density of expression of transmembrane TNF and is less pronounced for soluble receptor as compared to antibodies due to lower binding affinity. Induction of apoptosis is not essential for clinical efficacy in RA because all available TNF antagonists have the same efficacy profile, but this might explain differences in efficacy observed in distinct diseases.
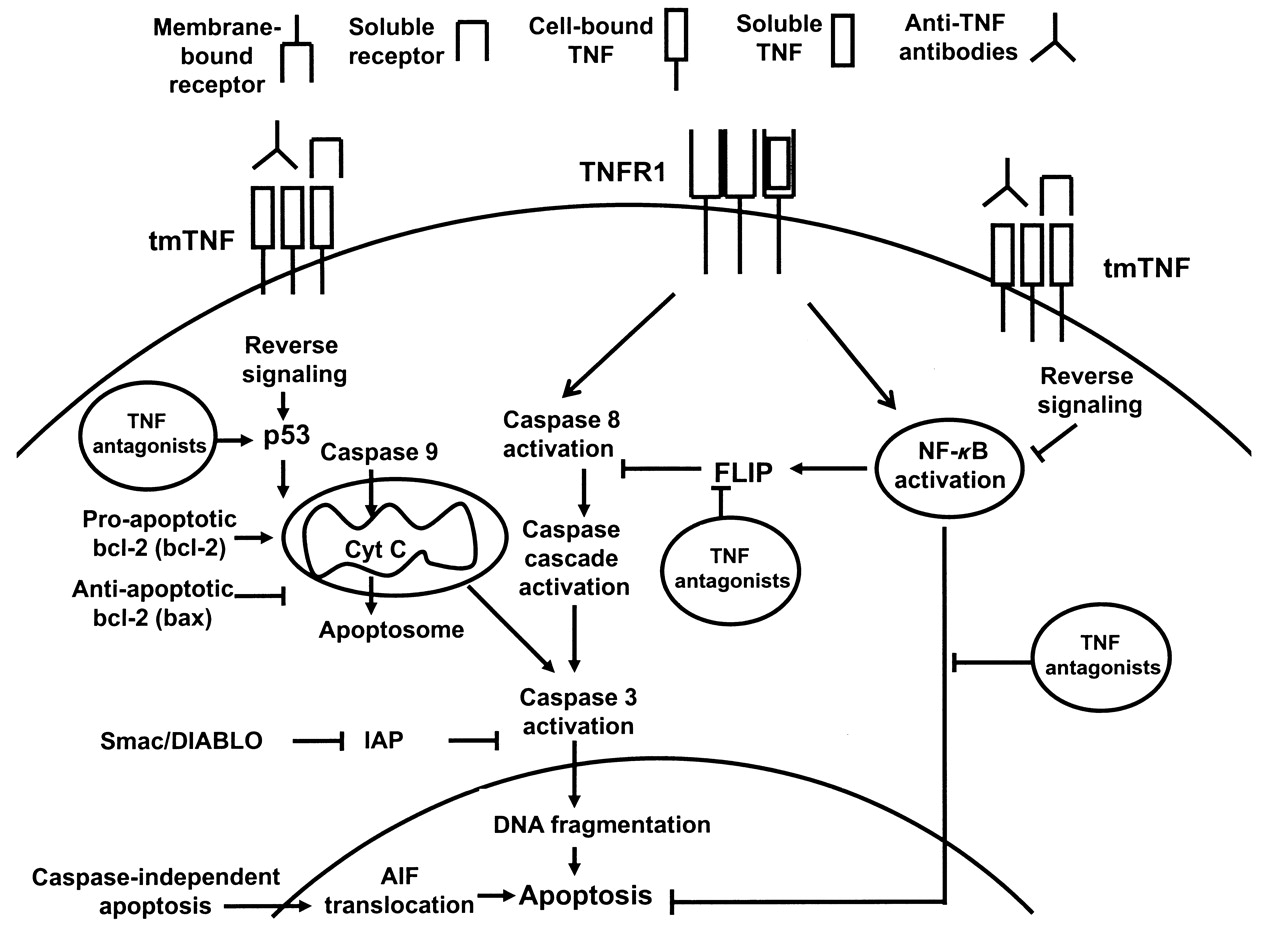
A representation of the apoptotic process with a focus on potential proapoptotic signals induced by tumor necrosis factor (TNF) antagonists in rheumatoid arthritis. TNFR: TNF receptor; tmTNF: transmembrane TNF; NF-κB: nuclear factor-κB; Cyt c: cytocrome c; IAP: inhibitors of apoptosis; AIF: apoptosis-inducing factor; FLIP: Fas-associated death domain-like interleukin 1ß-converting enzyme inhibitory protein; Smac/DIABLO: second mitochondria-derived activator of caspase/direct IAP-binding protein with low pI. Symbol legend given at the top.
Footnotes
-
Supported in part by research funding from the European Community FP7 funding project Gums and Joints, the Innovative Medicine Initiative Be the CURE, and the Swedish Research Council, and through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet and an unrestricted research grant from Centocor.
- Accepted for publication November 17, 2011.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵








{kind=link}