

Abstract
Objective. We assessed the role of Ank in the maintenance of postnatal articular cartilage using the ank/ank mouse (mice homozygous for progressive ankylosis).
Methods. We analyzed ank/ank mice and wild-type littermates (8, 12, and 18 weeks old). Sections from decalcified, paraffin-embedded joints were stained with hematoxylin and eosin. Articular chondrocyte size and cartilage thickness were determined using morphometric methods. Immuno-histochemical staining was performed with anticollagen X, antitissue nonspecific alkaline phosphatase (TNAP), and anti-ß-catenin antibodies on fixed joint sections. Axin2 expression in paw joint lysates in wild-type versus ank/ank mice were compared using Western blot analysis.
Results. In all age groups of normal mice studied, calcified cartilage (CC) chondrocyte areas were significantly larger than those of uncalcified cartilage (UC) chondrocytes. However, similar chondrocyte areas (UC vs CC) were found in 12-week and 18-week-old ank/ank mice, indicating that hypertrophic chondrocytes were present in the UC of these mutant mice. The ank/ank mice showed an increase in CC thickness. The ank/ank UC hypertrophic chondrocytes showed diffuse immuno-reactivity for collagen X and TNAP. Increased ß-catenin activation was demonstrated by nuclear localization of ß-catenin staining in ank/ank chondrocytes. Axin2 expression from paw lysates was downregulated in ank/ank mice.
Conclusion. We identified a previously unrecognized phenotype in the articular cartilage of ank/ank mice: collagen X-positive hypertrophic chondrocytes in the UC. It is possible that consequent to downregulation of axin2 expression, ß-catenin signaling was activated, leading to accelerated chondrocyte maturation and eventual ankylosis in ank/ank joints. Our studies shed new light on the contribution of a key signaling pathway in this model of joint ankylosis.
Inorganic pyrophosphate (PPi) metabolism is common to all cells and plays an important role in regulating mineralization and bone formation. PPi in the cartilage extracellular matrix (ECM) is exported by the Ank/ANKH protein1 and generated by ectonucleotidases, mainly ENPP-12,3. The mouse mutant Ank (progressive ankylosis) has a loss-of-function mutation in the Ank gene1. Although Ank is expressed in embryonic cells, ank/ank mice are normal at birth. It is possible that ENPP-1 compensates for Ank functions in embryonic development and early postnatal state. Adult ank/ank mice have pathologic calcium apatite crystal deposition and eventual bony ankylosis of the affected joints4. Mice with joint-specific deletion of Ank alleles also show joint mineralization and ankylosis5, indicating that Ank functions locally in joints. In fact, Ank gene expression is at least 16-fold higher in surface articular chondrocytes compared to the level expressed in growth plate chondrocytes6, suggesting that Ank has important functions in articular cartilage. The best documented function of Ank relates to the regulation of intracellular and extracellular PPi levels1,7. Other functions of Ank include regulating early erythroid8 and osteoblast/osteoclast differentiation9,10.
Modulators of Ank expression have been reported. For example, Ank expression is lower in hypoxic environments such as growth plate and articular cartilage, while higher expression has been found in the synovium and meniscus with normoxic conditions11. In rat chondrocytes, transforming growth factor-ß (TGF ß) induced Ank expression through the Ras/Raf-1/ERK and calcium-dependent protein kinase C pathways, leading to PPi export to the extracellular matrix12.
In view of the fact that Ank mice are outbred and our colony of Ank mice originated from a breeding pair derived from frozen embryos, we characterized the phenotypes of our ank/ank mice. We report the detection of a previously unrecognized phenotype in the articular cartilage of ank/ank mice, namely the presence of hypertrophic chondrocytes in the uncalcified cartilage (UC). We show that ß-catenin signaling is involved in the development of this phenotype.
MATERIALS AND METHODS
Ank mice
A breeding pair of heterozygous ank mice (C3FeB6-A/Aw−j - ank+/−) was purchased from Jackson Laboratory (Bar Harbor, ME, USA). Mice were genotyped using tail DNA as described by Ho, et al1. More than 50 mutant mice were evaluated. For histomorphometry, 4 mutant female (ank/ank) mice and 4 normal female littermate controls were analyzed for each age group (8, 12, and 18 weeks, except for 3 ank/ank mice who were 18 weeks old). Female mice were used exclusively in our study because the clinical phenotype in them is more severe than in male mice in our colony. All animal procedures were approved by the institutional Animal Experimentation Committee.
Histology and cartilage morphometry
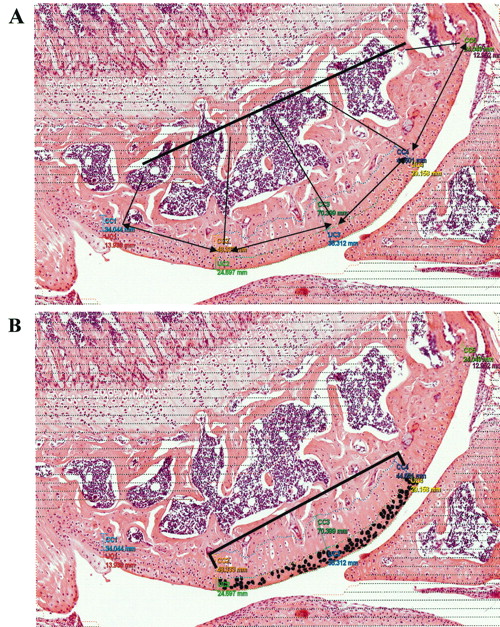
Decalcified fixed knee joint sections (5 μm) were stained with H&E. A Visiopharm Integrator System (Visiopharm Inc., Hoersholm, Denmark) was used for morphometry. For each mouse, the UC and calcified cartilage (CC) thickness of 1 knee joint was measured at 5 equidistant points across the area of interest. UC thickness was measured from the cartilage just beside the synovial space to the tidemark. CC thickness was measured from the tidemark to the edge of the subchondral bone. The mean UC/CC thickness of each mouse was used for statistical analyses. Within the cartilage from the 3 midpoints, chondrons with an intact nucleus, cytoplasm, and lacunae were evaluated. Each chondron was individually masked to obtain chondrocyte area by imaging (Figure 1). All the chondrocytes in the central 50% of the tibial plateau (∼ 200 chondrons) from each of the UC or CC for each mouse were measured. The mean chondrocyte area (from UC or CC) of each mouse was used for statistical analyses.
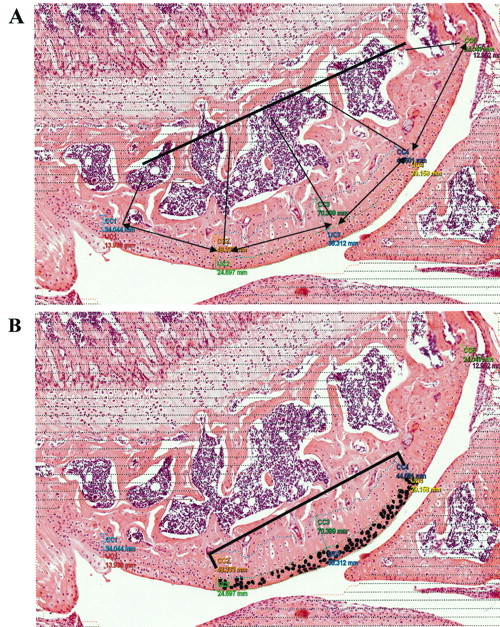
Histomorphometric analysis of knee joints using a Visiopharm Integrator System. A. The thickness of the uncalcified and calcified cartilage was measured at 5 equidistant points across the area of interest. B. Chondrons showing an intact nucleus, cytoplasm, and lacunae were evaluated in the central 50% of uncalcified and calcified cartilage. Each chondrocyte was individually masked (for example, blackened cells represent uncalcified cartilage chondrocytes) and image analysis was performed to obtain chondrocyte area.
Immunohistochemistry
Sections were treated with pepsin for antigen retrieval and 3% hydrogen peroxide to block endogenous peroxidase. The sections were stained either with anticollagen X (at 1:125 and 1:250 dilutions; Ibex Pharmaceuticals Inc., Montreal, QC, Canada) or with antitissue nonspecific alkaline phosphatase (TNAP; at 1:10 dilution) or with anti-ß-catenin (at 1:400 dilution) antibodies. MM-horseradish peroxidase (HRP) polymer was used as a detection system (BioCare-UK, Birmingham, UK), visualized with diamino-benzidine methods (Vectastain DAB Kit, Vector Laboratories, Burlingame, CA, USA). Sections were counterstained with Mayer’s hematoxylin. Positive and negative controls were stained in parallel. Specificity of the staining was verified each time using the same isotypic antibody as a control. For assessing the percentage of articular chondrocytes with nuclear localization of ß-catenin, ∼ 200 chondrocytes from 1 knee joint of each mouse (four 14-week-old mice each from both wild-type and ank/ank mice) were counted.
Immunoblotting
Skin from front paws of mice was removed. Interphalangeal joints from the front paws were solubilized in 6 M urea. Lysates with sodium dodecyl sulfate (SDS) and dithiothreitol were boiled for 3 min, loaded and run on SDS-PAGE, and transferred to Immobilon-P transfer membrane. After blocking with 3% bovine serum albumin in Tris-buffered saline with Tween 20 (10 mM Tris pH8, 500 mM NaCl, 0.1% Tween 20) for 1 h, the blots were incubated with either a mouse monoclonal antibody to Escherichia coli ß-galactosidase (ß-gal) or a goat anti-axin2 antibody (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA) or a monoclonal anti-ß-actin antibody for 45 min and then with HRP-conjugated antigoat or antimouse antibody (Jackson) for 30 min. Specific signals were detected by chemiluminescence using Supersignal West Femto maximum sensitivity substrates (Pierce Biotech, Rockford, IL, USA) and imaging.
Statistical analysis
Data from the different groups (4 mice in each group and three 18-week ank/ank mice) were compared using the nonparametric Mann-Whitney U test (SPSS v 16.0). P values < 0.05 were considered significant. Results were expressed as mean with 95% CI and were presented using box plots.
Generation of TOPGAL ank/ank mice
TOPGAL mice were purchased from Jackson Laboratory. Heterozygous Ank mice (homozygous mutants are infertile) were crossed with TOPGAL mice bearing the ß-gal transgene driven by a T cell factor (TCF) ß-catenin responsive promoter. F1 pro genies bearing the transgene were intercrossed to generate TOPGAL ank/ank mice. Genotyping for the transgene was carried out as described in Hens, et al13. Ank/ank mice with and without the transgene exhibit similar phenotypes (data not shown).
RESULTS
Radiology and histopathology
Progressive radiographic changes were detected in the menisci and the patella of ank/ank knees (Figure 2B). Axial joint abnormalities included syndesmophytes at costosternal junctions and extensive marginal syndesmophyte formation (Figure 2C, arrows) between vertebral bodies (“bamboo” spine). Histologically, these syndesmophytes were represented by proliferating osteoblasts and differentiated osteocytes in a mature bone matrix (Figure 2D, thick arrow). Calcium hydroxyapatite crystals were present in the intervertebral space (Figure 2D, dashed arrow). A large number of hypertrophic chondrocytic-like cells were detected in the proximity of new bone formation (Figure 2D, thin arrow). No inflammation was observed.

Radiographic and histological evaluation of joints from wild-type (wt) and ank/ank mice. A. Radiograph showing knee joint of a wt mouse (12 weeks). B. Radiograph showing knee joint of an ank/ank mouse (18 weeks). Arrow shows extensive calcification and complete knee joint ankylosis. C. Radiograph of ank/ank spine (12 weeks) showing bridges of new bone formation (syndesmophytes) extending from 1 vertebral edge to another (arrows). D. Histological section of ank/ank syndesmophyte, showing bridges of mature bone formation (thick arrow) and hydroxyapatite crystals (dashed arrow). Hypertrophic chondrocytes are identified in the vicinity of new bone formation areas (thin arrow). E. A higher magnification image of an ank/ank syndesmophyte (SYN) showing proliferating osteoblasts (arrows). GP: growth plate.
Morphometry of chondrocytes in the knee joints
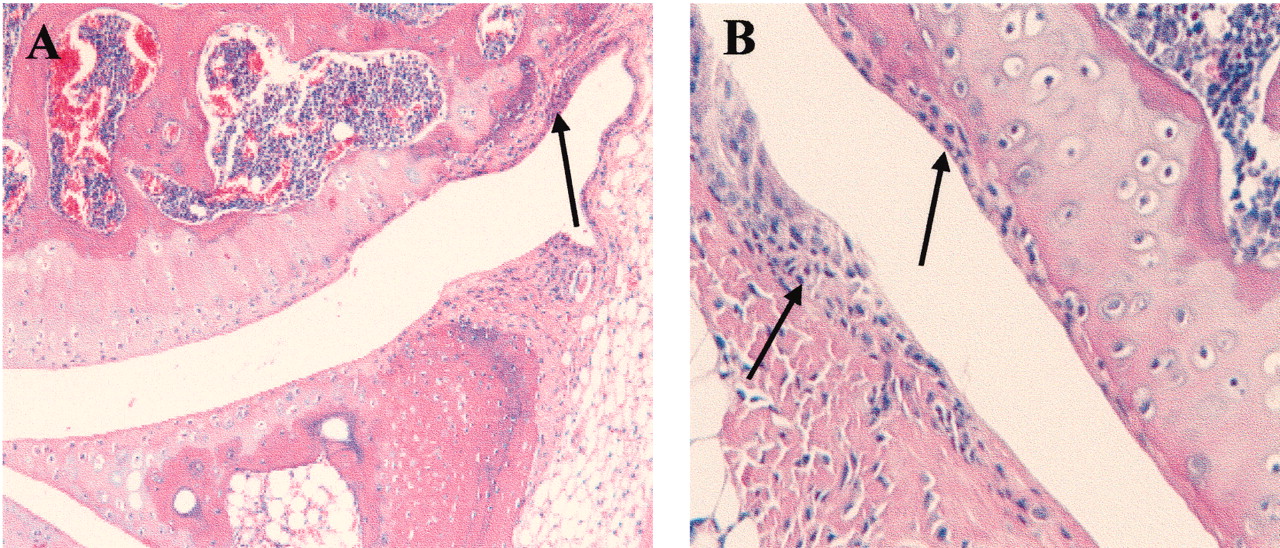
As observed by Sampson14, the peripheral joints of ank/ank mice showed proliferation of the fibroblast-like synoviocytes (Figures 3A and 3B; arrows). In contrast to previous reports showing a mononuclear inflammatory infiltrate in synovia4, no inflammation was seen in our ank/ank mice at any age.
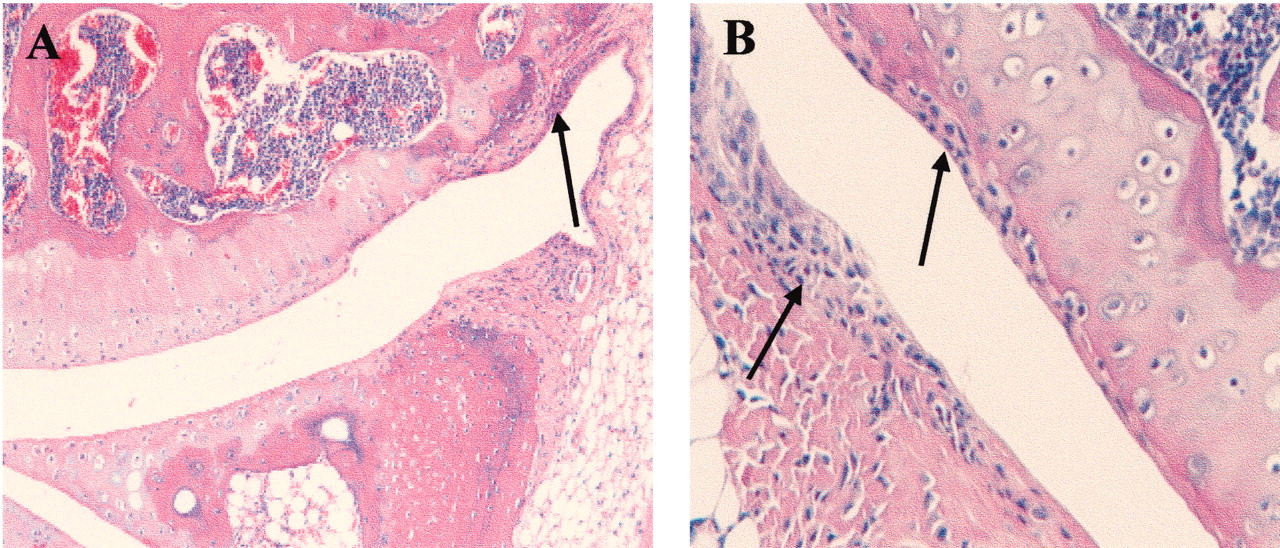
Histological features of ank/ank articular joints. A. The cartilage layer is intact with no signs of infiltration in the joint space. Arrow shows proliferating fibroblast-like synoviocytes. B. A higher-power image showing the absence of leukocyte infiltrates in synovial hyperplasia (arrows).
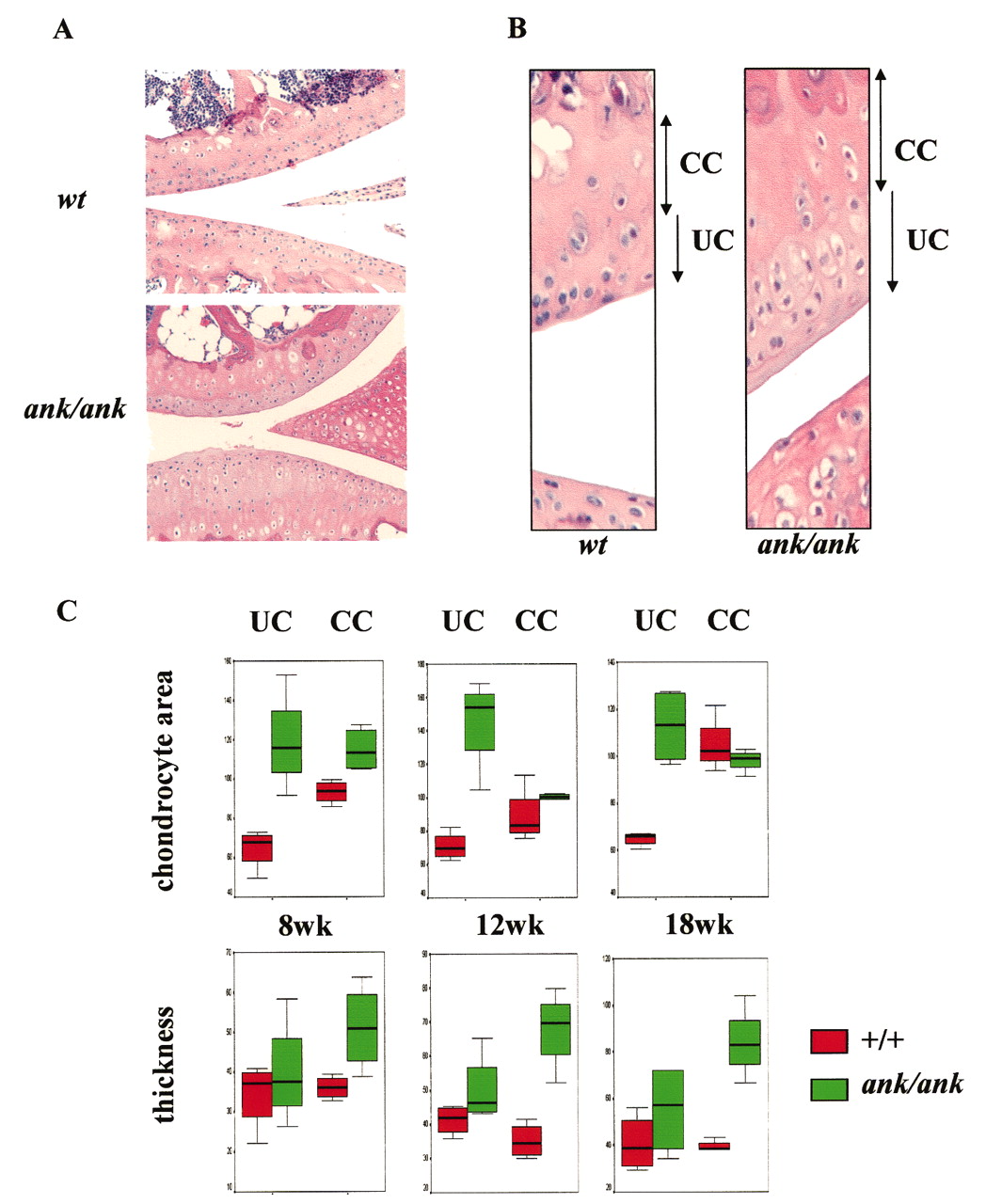
Detailed examination of the UC revealed chondrocyte hypertrophy in ank/ank mice compared with wild-type controls (Figures 4A and 4B). Normally, hypertrophic chondrocytes are never found in UC and thus one would expect a significant difference in the UC versus CC chondrocyte area in the articular cartilage of normal mice. Indeed, in all 3 age groups of normal mice, average CC chondrocyte areas were significantly larger than those of UC chondrocytes (p = 0.021 for all age groups). Significant differences in average chondrocyte areas were also detected in 8-week ank/ank UC versus CC (p = 0.021). However, similar mean chondrocyte areas (UC vs CC) were found in 12-week and 18-week ank/ank articular cartilage (p values were nonsignificant), indicating that hypertrophic chondrocytes were present in the articular UC of 12-week and 18-week ank/ank mice (Figure 4C).

Articular cartilage from wild-type (wt) versus ank/ank mice. A. Histological evaluation of articular cartilage from wt versus ank/ank mice (both 18 weeks of age). Ank/ank joints showed joint space narrowing and amorphous debris within the joint space. B. Detailed histological features of articular cartilage from the knee joint of wt versus ank/ank mice (both 12 weeks of age). UC: uncalcified cartilage; CC: calcified cartilage. C. Box plots comparing UC versus CC chondrocyte area (top panels), UC versus CC thickness (bottom panels) of wt mice (red boxes) versus ank/ank mice (green boxes) at ages 8, 12, and 18 weeks.
There was a progressive increase in the CC thickness in ank/ank mice, compared to that of wild-type mice. In wild-type articular cartilage, there was no significant difference between the UC versus CC thickness in all 3 age groups. In contrast, in all 3 age groups of ank/ank articular cartilage, CC thickness was significantly greater than that of UC (Figure 4C).
Immunohistochemical (IHC) studies of the knee joints
In wild-type mice, collagen X expression, a marker for hypertrophic chondrocytes, was detected primarily in the hypertrophic chondrocytes in the articular CC (Figure 5A), and was not detected in the uncalcified chondrocytes. In contrast, chondrocytes from the UC of ank/ank mice showed membrane and cytoplasmic reactivity for collagen X, and hypertrophic chondrocytes from the CC were also positive (Figure 5B). A semiquantitative analysis of collagen X positivity, using 2 different antibody concentrations, indicated that collagen X protein expression was more than 2-fold higher in the articular hypertrophic chondrocytes from ank/ank mice, compared to wild-type mice.

Immunohistochemical staining of collagen X (Col X) and tissue nonspecific alkaline phosphatase (TNAP) of wild-type (wt) versus ank/ank articular joint sections. A. The wt joint section was incubated with anticollagen X antibody at 1:125 dilution. B. The ank/ank joint section was incubated with the same antibody at 1:250 dilution. C. The wt joint section was stained with anti-TNAP antibody. Only focal chondrocytes from the calcified cartilage showed positive staining. D. The ank/ank joint section was stained with anti-TNAP antibody. Accentuated TNAP staining was detected in both uncalcified and calcified articular chondrocytes.
TNAP has long been regarded as a marker of chondrocyte hypertrophy15. We recently showed that dysregulation of Ank gene expression affected TNAP expression and activities16. Thus, we carried out IHC studies of the ank/ank versus normal knee joints using an anti-TNAP antibody. TNAP staining was consistently higher in the articular hypertrophic chondrocytes from the ank/ank UC (Figure 5D). As expected, normal articular cartilage had only focal TNAP positivity in the uncalcified layer (Figure 5C).
Activation of ß-catenin signaling in ank/ank chondrocytes
Canonical wingless (Wnt)/ß-catenin signaling is involved in bone formation remodeling during growth and development and in the postnatal state17. Activation of this signaling pathway in mature chondrocytes stimulates hypertrophy and matrix mineralization18. Ank/ank mice and ß-catenin conditional activation (cAct) mice19 share a common hypertrophic chondrocyte phenotype in the postnatal articular cartilage. Thus we asked whether ß-catenin signaling is dys-regulated in ank/ank mice. IHC studies of ank/ank versus normal knee joints using an anti-ß-catenin antibody showed nuclear localization of ß-catenin (a marker for activation of ß-catenin signaling) in ank/ank articular chondrocytes. In young normal mice (up to 8 weeks of age), ß-catenin staining was observed in the cytoplasm and membrane of articular cartilage (Figure 6A). In contrast, in young ank/ank mice, intense ß-catenin expression (mainly localized in the nuclei) was detected in both articular and epiphyseal chondrocytes (Figure 6B and 6D). Strong ß-catenin staining was present in the epiphyseal intercellular matrix, predominantly in the proliferating and prehypertrophic zones (Figure 6D, arrow). Weak ß-catenin staining was observed in the epiphyseal cartilage matrix from young normal mice (Figure 6C). High expression of ß-catenin was also present in the perichondrium of ank/ank mice (Figure 6B, arrow).

Immunohistochemical staining of ß-catenin in articular and epiphyseal cartilage of wild-type (wt) versus ank/ank mice (both 8 weeks old). A. Weak cytoplasmic and membrane staining of the articular cartilage of wt mice. B. Strong ß-catenin staining in ank/ank articular chondrocytes with nuclear localization. Arrow shows higher expression of ß-catenin in the ank/ank perichondrium. C. Weak cytoplasmic and membrane staining (arrow) of the epiphyseal cartilage of wt mice. D. Prominent ß-catenin staining in ank/ank epiphyseal cartilage, mainly in the proliferating and prehypertrophic zones (arrow) and the epiphyseal intercellular matrix.
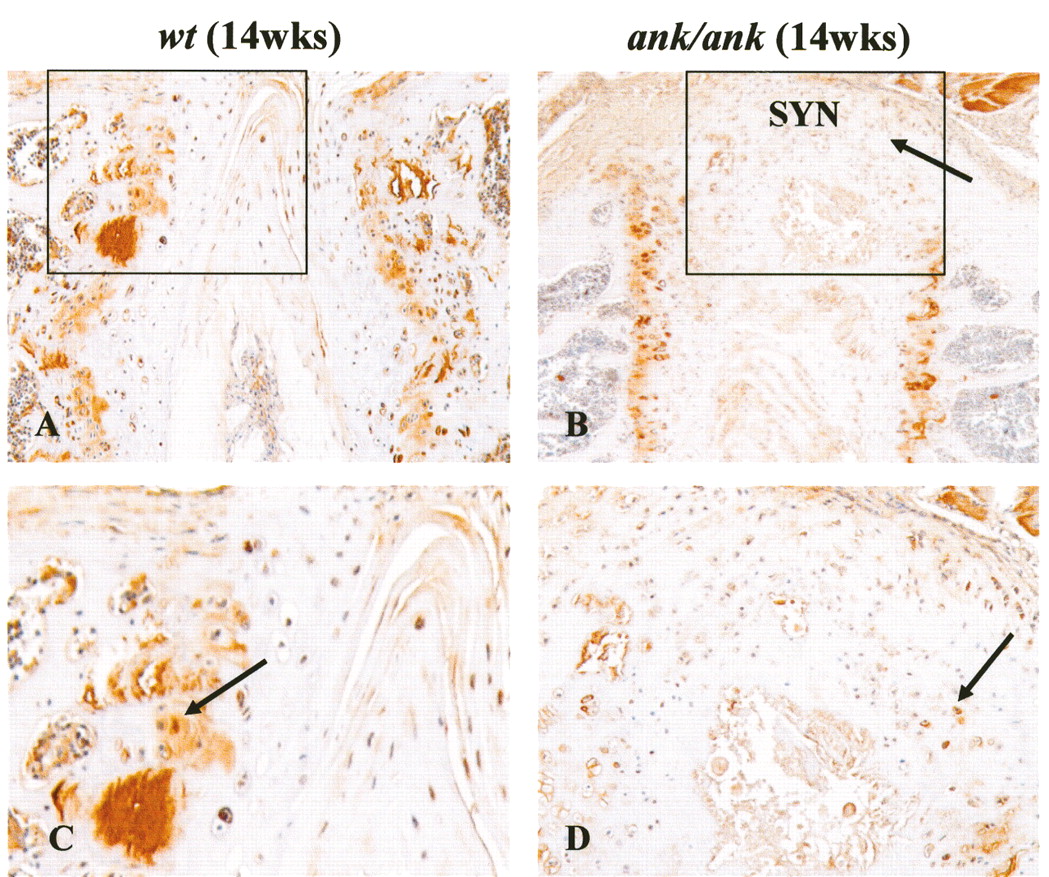
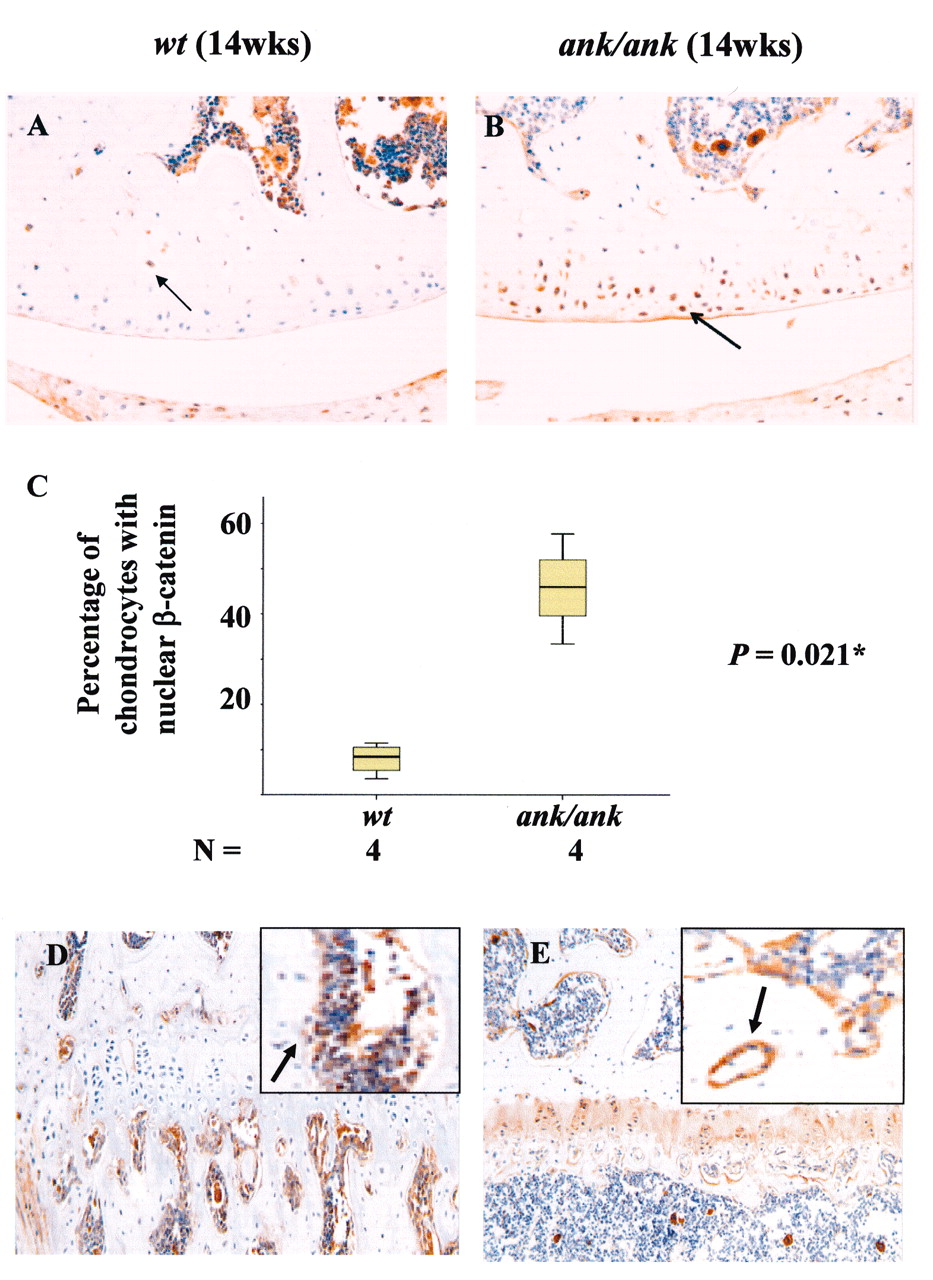
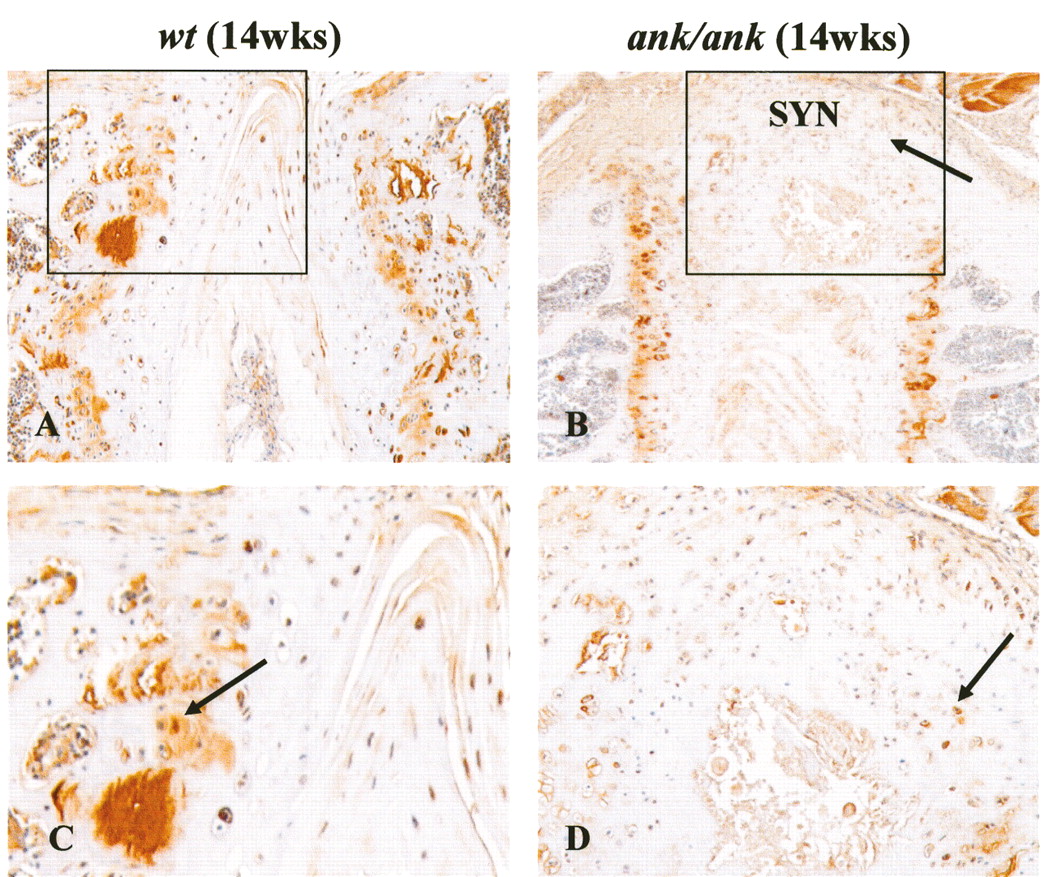
In mature normal mice (12–17 weeks old), little or no ß-catenin staining was detected in the superficial layer of articular chondrocytes, epiphyseal chondrocytes, matrix, or perichondrial cells (Figures 7A and 7D). Focal positive staining was observed only in the deep layer of chondrocytes in the articular cartilage (Figure 7A) and the trabecular bone (Figure 7D). In contrast, strong cytoplasmic and nuclear ß-catenin staining persisted in all layers of the articular cartilage from mature ank/ank mice (Figure 7B). In 14-week-old normal mice, 8% of articular chondrocytes (2.6–13.4%; n = 4 mice; UC chondrocytes counted from 1 knee joint of each mouse) showed nuclear localization of ß-catenin. In age-matched ank/ank mice, nuclear staining was found in a significantly higher percentage of articular chondrocytes (46%; 30.1–61.6%; n = 4 mice; UC chondrocytes counted from 1 knee joint of each mouse; p = 0.021; Figure 7C). Similar intense staining was present in the epiphyseal chondrocytes, cartilage matrix, and in perichondrial cells of ank/ank mice (Figure 7E). Syndesmophytes from the mature ank/ank spine showed intense ß-catenin staining in osteoblasts and in the chondrocytic-like cells in the vicinity of new bone formation (Figures 8B and 8D). Positive ß-catenin staining was present only in the CC of the vertebral bodies from the normal spine.

Immunohistochemical staining of ß-catenin in articular and epiphyseal cartilage of wild-type (wt) versus ank/ank mice (both 14 weeks old). A. In mature wt joint sections, little or no ß-catenin staining (arrow) in the superficial layers of articular chondrocytes. B. Mature ank/ank articular cartilage showed strong cytoplasmic and nuclear staining (arrow). C. Box plot comparing the percentage of articular chondrocytes with nuclear ß-catenin staining in wt versus ank/ank knee joints (14 weeks old, n = 4 for both groups). Mann-Whitney U test showed significant differences (*p = 0.021). D. No ß-catenin staining observed in the epiphyseal chondrocytes from mature wt mice. Only focal staining was observed in the trabecular bone (arrow in insert). E. Marked staining was present in the epiphyseal chondrocytes and cartilage matrix of mature ank/ank mice. B-catenin staining was detected in osteoblasts and newly formed osteoid in the trabecular bone from mature ank/ank mice (arrow in insert).
Immunohistochemical staining of ß-catenin in spinal articular cartilage of wild-type (wt) versus ank/ank mice (both 14 weeks old). A. and C. Positive staining is present mainly in deep articular cartilage from wt mice (arrow). B. and D. In ank/ank mice, in addition to strong signals in the growth plates, strong nuclear and cytoplasmic ß-catenin staining is detected in syndesmophytes (SYN) and areas of new bone formation associated with chondrocyte hypertrophy (arrows). Panels C and D are higher magnification images from inset areas of panels A and B, respectively.
We unexpectedly observed positive ß-catenin staining in the ECM, especially in the growth plates (more prominent in the mutant than normal mice). Although little staining was detected in negative control sections, we cannot rule out the possibility that this finding might reflect some degree of cross-reactivity of the primary antibody used. To confirm that ß-catenin signaling was upregulated in ank/ank joints using an alternative approach, we generated ank/ank mice with a ß-gal transgene (TOPGAL) driven by a TCF ß-catenin responsive promoter13. Western blots of front paw joint lysates from TOPGAL-positive versus TOPGAL ank/ank mice (two 12-week mice each) were probed with an antibody to bacterial ß-gal, and ß-actin expression was used as a normalization marker. Higher ß-gal signals were detected in lysates from TOPGAL ank/ank mice (Figure 9A).
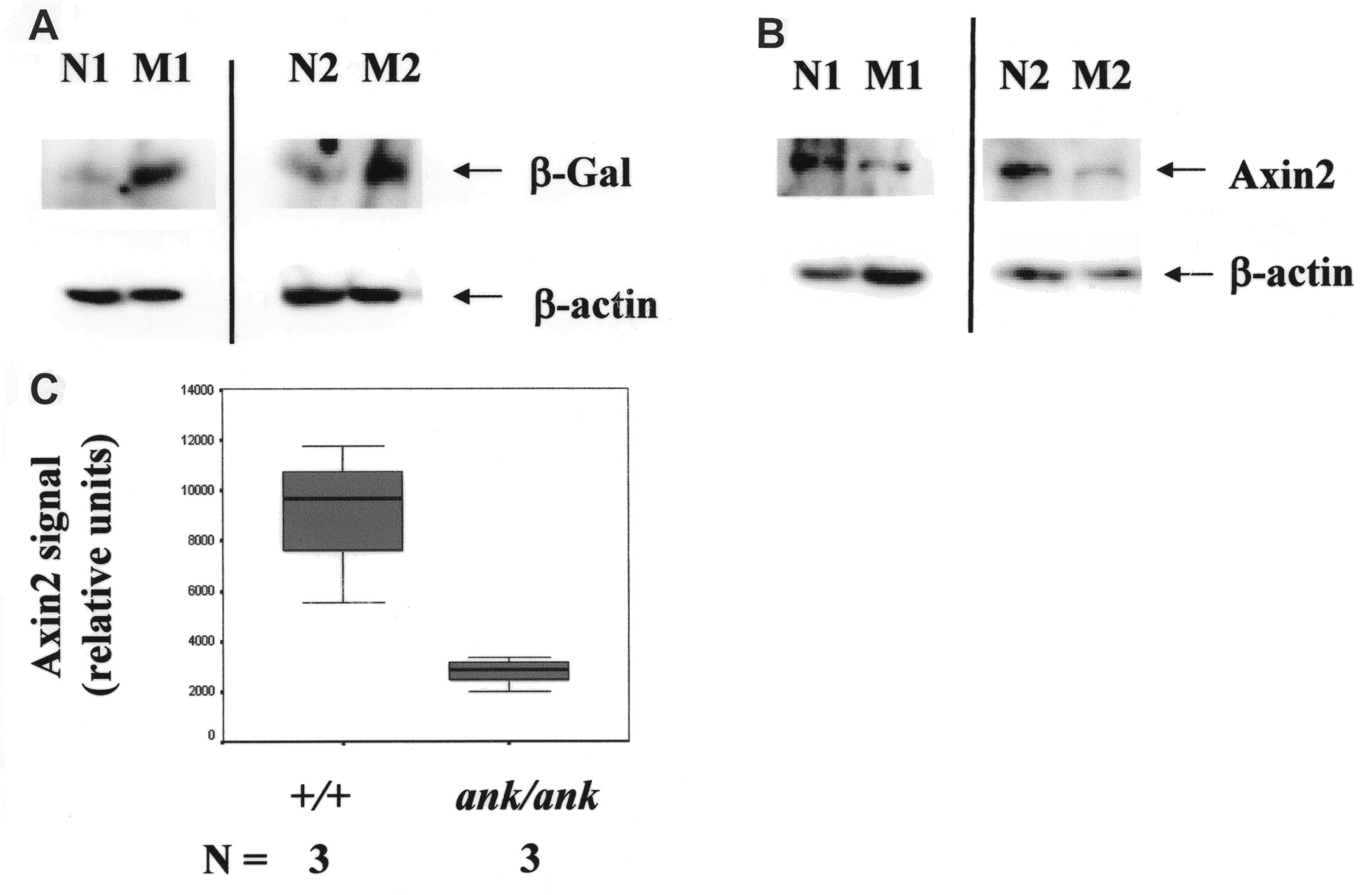
Western blot analyses. A. Paw joint lysates [TOPGAL wild-type (N1 and N2) vs TOPGAL ank/ank (M1 and M2)]. N1 and M1 and N2 and M2 are from the same litter, respectively. Blots in the top panels were probed with an antibody to bacterial ß-gal and blots in the bottom panels were probed with an antibody to ß-actin (used for normalization of protein loading). B. Representative Western blot of paw joint lysates probed with an antibody to axin2. Results of lysates from 2 pairs of wild-type (N1 and N2) versus ank/ank (M1 and M2) mice are shown. ß-actin expression was used as a normalization marker. C. Quantitation of relative levels of axin2 expression. Significantly lower axin2 levels were detected in ank/ank joints (n = 3 mice for each group; p = 0.05).
Role of axin2 in the development of the hypertrophic chondrocyte phenotype in ank/ank mice
Axin2 is a negative regulator of canonical Wnt/ß-catenin signaling20. It is also a concentration-limiting factor in the ß-catenin degradation complex21,22. In view of the fact that disruption of axin2 expression resulted in accelerated chondrocyte maturation23, we assessed whether axin2 expression was dysregulated in ank/ank joints. Because posttranscriptional regulation plays an important role in proteins that participate in ß-catenin signaling, we compared the levels of axin2 proteins in wild-type versus ank/ank joint lysates. Western blots of front paw joint lysates were probed with an antibody to axin2, and ß-actin expression was used as a normalization marker. Compared to those from normal littermates, there were 2- to 5-fold fewer axin2 proteins in joint lysates from ank/ank mice. Figure 9B shows representative results from 2 pairs of normal versus mutant mice. Quantitation of the relative levels of axin2 expression is shown in Figure 9C (n = 3 mice per group; p = 0.05).
DISCUSSION
In our study, we report a previously unrecognized phenotype in ank/ank mice, namely hypertrophic chondrocytes in the UC, associated with an increase in CC thickness. Although hypertrophic chondrocytes were detected in both UC and CC of ank/ank articular cartilage, there was no increase in the UC thickness, while CC thickness increased progressively with age. It is likely that the hypertrophic chondrocytes in the UC of ank/ank mice were not terminally differentiated, or inhibitory factors were present and thus ECM mineralization did not occur in the UC. We have also shown that ß-catenin signaling is activated in ank/ank articular cartilage and that lower axin2 protein levels are detected in ank/ank joint lysates. A recent study by Cailotto, et al showed that silencing Ank in rat resting articular chondrocytes mimicked the effect of interleukin 1ß on chondrocyte phenotype markers (lack of collagen X, TNAP, and runx-2 expression)24. Further, Wnt5a transcript was increased about 2-fold and is likely responsible for activation of the canonical Wnt-ß-catenin signaling in chondrocyte monolayer. This is an ePPi-dependent event, because exogenous PPi compensates for the Ank-silencing effects18.
There are a few limitations in the Cailotto study24. First, our observation of the hypertrophic chondrocytes in ank/ank articular joints suggests that resting articular chondrocytes likely are not the predominant cell population in vivo when Ank is deficient. Second, analysis of rat resting articular chondrocytes in vitro24 cannot address the consequence of activation of ß-catenin signaling in articular joints in vivo. Third, articular chondrocytes in vivo are nonproliferative and exist under hypoxic conditions. Thus, the in vitro systems used in the rat chondrocyte cultures24 differ from the in vivo conditions. This is highly relevant because Ank expression is repressed in hypoxic conditions, being regulated by hypoxia-inducible factor-111. In the ank/ank articular joints, it remains unclear whether there is upregulation of Wnt proteins. This is currently under investigation.
It is possible that in ank/ank joints, activation of ß-catenin signaling is consequent to lower levels of axin2, which is a negative regulator of Wnt signaling21. This possibility is supported by the fact that 3 mutant mice (ank/ank), axin2 deficient23, and ß-catenin conditional activation (cAct) mice19, shared a common accelerated chondrocyte maturation phenotype. It will be of interest to assess whether restoration of axin2 to its normal levels (for example, by stabilizing axin2 degradation) could modulate ß-catenin signaling in ank/ank chondrocytes.
It is unclear why axin2 expression is downregulated in ank/ank joints. Axin2 is involved in mediating the crosstalk of TGF-ß and Wnt/ß-catenin signaling25. There is evidence showing that Ank participates in TGF-ß signaling, the most compelling result being that PPi export (a known Ank function) was greatly diminished in TGF-ß-stimulated rat chondrocytes transfected with Ank small interfering RNA24. In postnatal mouse sternal chondrocytes, a shift from TGF-ß to Wnt/ß-catenin signaling leads to an acceleration of chondrocyte maturation25. This raises the possibility that aberrant TGF-ß signaling may contribute to activation of Wnt/ß-catenin signaling in ank/ank chondrocytes, leading to the hypertrophic chondrocyte phenotype and progressive ankylosis in the mutant mice. This notion is supported by the fact that similar to ank/ank mice, mice deficient in TGF-ß signaling26,27 have hypertrophic chondrocytes in the articular UC.
There are 2 distinct joint-related phenotypes of ank/ank mice that mimic 2 major human rheumatic diseases: hypertrophic chondrocytes in osteoarthritic articular cartilage and the joint ankylosis of ankylosing spondylitis (AS). Canonical Wnt/ß-catenin signaling has been implicated in the pathogenesis of osteoarthritis (OA)28. In contrast to the Ank-deficient hypertrophic chondrocytes found in articular cartilage of ank/ank mice, the hypertrophic chondrocytes seen in OA joints showed upregulation of ANKH expression29. It appears paradoxical that increased Ank expression was detected in hypertrophic chondrocytes from growth plates and osteoarthritic cartilage, while hypertrophic chondrocytes were also detected in ank/ank articular UC where there are no functional Ank proteins. For systems that have feedback, loss of function and gain of function could result in a similar phenotype. A similar situation was present in mouse mutants, in which both the loss and gain of function of ß-catenin in articular cartilage resulted in a similar OA-like phenotype, but the underlying mechanisms were different19,30.
Despite the presence of hypertrophic chondrocytes and activated ß-catenin signaling in ank/ank mice, unlike OA, our mutant mice do not demonstrate joint fibrillation or progressive degenerative joint disease. In prostate cancer metastatic to the bone, the outcome of the bone phenotype could be osteoinductive or osteolytic, and appeared to be determined by the presence/absence of bone morphogenetic protein and Wnt signaling antagonists31,32. It is tempting to hypothesize that dysregulation of these signaling antagonists leads to the development of ankylosis (rather than cartilage degradation) in ank/ank mice. In prior studies, both the presence of joint inflammation4 and the lack of evidence of inflammation in the ank/ank joints33 were reported. In our colony of ank/ank mice, there were no signs of joint inflammation, and this might explain the lack of joint destruction in our mutant mice. Unlike ank/ank articular cartilage, there is no increase in CC in patients with OA. These differences are expected, because OA is a complex disease involving numerous risk factors, while ank/ank mice have a single gene defect.
We and others showed previously that ANKH was modestly associated with AS34,35, although no ANKH association was detected in a UK study36. Even if ANKH polymorphisms do not constitute a risk factor for AS in genetic studies, it is possible that localized dysregulation of ANKH occurs in AS joints, because growth factor37 and androgen38 influence ANKH expression. Ank/ank mice represent an informative model for the study of joint ankylosis in AS for 3 reasons, as follows: (1) Progressive joint ankylosis, both peripheral and axial, is a phenotype shared by both the ank/ank mice and patients with AS. (2) We showed that Wnt/ß-catenin signaling is activated in ank/ank joints. There is increasing evidence that this signaling pathway plays an important role in joint ankylosis in patients with AS. A recent report showed that serum Dickkopf-1 (DKK-1; a Wnt antagonist) from patients with AS is dysfunctional and thus likely leads to neoossification in these patients through activation of the Wnt/ß-catenin signaling pathway39. In a murine model of sacroiliitis, blocking DKK-1 resulted in local chondrocyte hypertrophy, upregulation of type X collagen expression, and enhanced ankylosis of the sacroiliac joints40. The role of Wnt/ß-catenin signaling in the development of ankylosis in patients with AS is also implicated by the finding that expression of sclerostin (SOST), an antagonist of ß-catenin signaling, was undetectable in AS osteocytes41. And (3) the hallmark of AS is osteoproliferation at the site of inflammation. Although anti-tumor necrosis factor agents are extremely successful in controlling joint inflammation in AS, results of numerous studies have revealed that they have minimal effect on new syndesmophyte formation42,43. Currently, there is an active debate whether ankylosis and inflammation are uncoupled in AS44. The unique feature in our ank/ank mice (ankylosis in the absence of joint inflammation) offers an opportunity to directly separate the molecular basis underlying ankylosis in the absence of confounding factors due to joint inflammation. Identifying downstream events that are common in the ankylosing process of ank/ank mice will lead to potential therapeutic targets for the human disease. These mutant mice will provide an experimental platform that enables modeling and testing of novel antiankylosis treatment strategies. Further, understanding the underlying mechanisms of the ank/ank phenotypes would improve our knowledge of postnatal articular cartilage homeostasis and may shed new light on pathogenic events in rheumatic diseases associated with chondrocyte hypertrophy and pathological new bone formation.
Our study has some limitations. It remains unclear whether there were any abnormal changes in the expression of Wnt proteins (such as Wnt5a)/Wnt agonists (such as R-spondins) or Wnt antagonists (such as SOST and DKK-1) in ank/ank mice that might contribute to activation of ß-catenin signaling. An in-depth assessment is warranted to address this issue. In addition, it is important to investigate whether increased chondrocyte hypertrophy and activation of ß-catenin signaling is a direct or indirect effect of loss of Ank function in the mutant mice.
We detected 2 novel in vivo abnormalities relating to the progressive ankylosis phenotype of ank/ank mice: (1) hypertrophic chondrocytes are aberrantly present in ank/ank articular UC; and (2) ß-catenin signaling is activated in ank/ank joints as evidenced by nuclear localization of ß-catenin, and higher ß-gal signals in the paw joints of TOPGAL ank/ank mice, likely due to fewer axin2 proteins in ank/ank chondrocytes. Our findings suggest that aberrant chondrocyte maturation and activation of ß-catenin signaling underlie joint ankylosis in ank/ank mice.
Acknowledgment
We are thankful for the skilled technical assistance of Gordana Kuruzar and Muntajib Alhaq, MLT, for the histological preparation, and Ruoyu Ni, BMed, MLT, for his assistance with immunohistochemistry, and Eric Gracey for photography.
Footnotes
-
Dr. F.W.L. Tsui and Dr. R.D. Inman are both senior authors of this report.
-
Supported by grants from the Canadian Institutes of Health Research, the Arthritis Center of Excellence, The Arthritis Society, and Pathology and Laboratory Medicine, Mount Sinai Hospital.
- Accepted for publication October 26, 2011.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}