

Abstract
Objective. To describe clinical phenotypes in neuropsychiatric systemic lupus erythematosus (NPSLE).
Methods. Data were prospectively collected in the Leiden NPSLE referral clinic, where patients suspected of having NPSLE are assessed in a standardized multidisciplinary manner. In consensus meetings, all medical specialists agreed on therapeutic strategy based on the suspected pathogenetic mechanism of NPSLE in the individual patient. An algorithm illustrates the process of decision-making during the consensus meeting. Clinical phenotypes are described, classified by pathogenetic mechanism.
Results. One hundred consecutive patients were evaluated, of whom 71 had SLE (29 patients did not fulfill ≥ 4 American College of Rheumatology criteria) and 46 had NPSLE. Primary NPSLE was diagnosed in 38 patients (53%) and could be differentiated in 21 patients (55%) with inflammatory NPSLE who were advised on immunosuppressive therapy, 12 patients (32%) with ischemic NPSLE who were advised on anticoagulant therapy, and 5 patients (13%) with undefined NPSLE who were advised symptomatic treatment only. Cognitive dysfunction and higher level of disease activity were associated with inflammatory NPSLE. Although presence of immunoglobulin G anticardiolipin antibodies and abnormalities on magnetic resonance imaging (MRI) were associated with ischemic NPSLE, abnormalities on MRI lacked specificity to distinguish phenotypes. A history of renal disease and use of corticosteroids were associated with secondary NPSLE.
Conclusion. We describe multidisciplinary consensus as a standard for diagnosing and defining phenotypes in NPSLE. These phenotypes show specific characteristics, which can be used to support diagnosis and guide therapeutic decisions. Clinical phenotyping and selection of patients becomes increasingly important when advances in experimental science lead to new targets for therapy in NPSLE.
Understanding of pathogenesis in neuropsychiatric systemic lupus erythematosus (NPSLE) is emerging and recent experimental work links autoantibodies to cognitive dysfunction1,2. Cognitive dysfunction is reported in up to 80% of patients with SLE, but it can also be a nonspecific finding3,4. Uniformity and transparency in establishing NPSLE has improved since the introduction of the 1999 American College of Rheumatology (ACR) Nomenclature and case definitions, but the usefulness in clinical practice is limited5,6,7. Therefore, NPSLE still presents a challenge to the clinician and usually involves the expertise of several medical specialists.
To date, therapeutic decisions in NPSLE are made per individual patient and are based on the suspected pathogenetic cause and severity of symptoms8,9. Proposed etiological mechanisms in primary NPSLE are inflammation, cytokine- or autoantibody-mediated neuronal dysfunction or damage, intracranial angiopathy, and ischemia and thrombotic events10,11. Therapy can be directed at inflammation with immunosuppressive medication or at ischemia and thrombotic events with anticoagulants. Further, especially in mild cases, therapy can focus on symptoms and consist of antidepressants, anticonvulsants, or antipsychotics only. In addition, in secondary NPSLE, patients have various neuropsychiatric (NP) symptoms due to the medication for SLE or to SLE-related organ damage.
Evidence for the selection of patients for immunosuppressive therapy, e.g., cyclophosphamide, is largely lacking, as are data on the phenomenology of NPSLE per pathogenetic cause12,13. The goal of our study was to describe in detail the multidisciplinary diagnostic approach and different clinical phenotypes of patients with NPSLE. Clinical phenotypes are based upon the suspected pathogenetic mechanism and include inflammatory NPSLE, ischemic NPSLE, undefined NPSLE, and secondary NPSLE.
The Leiden University Medical Center serves as a tertiary referral center for NPSLE. Because of the limited availability of standardized prospective data14, we started the Leiden NPSLE clinic in 2007 to evaluate patients with a suspicion of NPSLE in a standardized, multidisciplinary way. This tertiary care facility aids physicians in diagnosing and treating NPSLE, leading to a prospectively collected database. We used this database of patients with NPSLE to describe the phenotypes.
MATERIALS AND METHODS
In order to diagnose NPSLE we designed a 1-day program for assessment of patients by all relevant medical specialists. Therapeutic decisions were made based on the suspected underlying pathogenetic mechanism of NPSLE by consensus of all participating medical specialists. Table 1 offers an outline of patient assessment.
Procedure in evaluation of patients.
Patients
All patients suspected of having NPSLE and who speak Dutch or English can be referred to the Leiden NPSLE clinic by their treating physician. Patients give written informed consent for the storage of clinical data including serum and DNA for future research purposes.
Sociodemographic variables were assessed for all patients, including age, education level (primary, low education, 0−8 years; secondary medium education, 9−16 years; and high vocational/university education), and ethnicity. A diagnosis of ≥ 4 ACR criteria15,16 was mandatory for the diagnosis of SLE.
Rheumatology assessments
Patients were assessed by a rheumatologist (GMS-B) for current signs and symptoms, use of medication, medical history, and family history. A general physical examination was performed. The assessment was specifically aimed at past and current manifestations of SLE disease activity and end-organ damage due to SLE. Patients are classified according to the revised ACR criteria for SLE15,16 and the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI)17. Disease duration and symptom duration is extracted from the medical records when possible or derived from the history.
Internal medicine
Patients were assessed by a resident in internal medicine (TRS) under the close supervision of an internist (MVH) specializing in vascular medicine for symptoms of former and current vascular diseases. Special attention was paid to symptoms of atherosclerotic disease, thrombotic events, vasculitis, and cardiovascular (CV) risk factors including hypertension, dyslipidemia, and diabetes. According to the SCORE system for management of CV risk, risk factors are denominated as follows: hypertension if systolic blood pressure is > 140 mm Hg, obesity if body mass index exceeds 29.9, hypercholesterolemia if total cholesterol exceeds 6.5 mmol/l18.
Neurology
A neurological assessment was done by an experienced neurologist (ELEMB) and focused on headache and signs of seizures, alertness, and motor and sensory deficits. Examination includes fundoscopy, examination of cranial nerves, visual fields, strength of arm muscles and dexterity, observation of gait and ataxia, walking on toes and heels, tendon reflexes, Babinski reflex, sensory examination of gnostic and vital abilities, fingertip-nose test, muscle tone, and muscle atrophy. An acute focal neurological deficit with resolution within 24 h is reported as a transient ischemic attack (TIA)5. If indicated, assessment of cerebrospinal fluid (CSF), electroencephalography, or electromyography are performed.
Psychiatry
Psychiatric assessment is done by a resident in psychiatry (EB) under close supervision of a psychiatrist (RCvdM or NJAvdW) and includes a detailed psychiatric history and mental status examination assessing behavior, cognition, perception, and thinking, as well as mood and affect in a standardized manner. The following 4 instruments are used to assess patients: the Medical Outcomes Study Short Form-36 (SF-36) for measurement of self-reported quality of life regarding physical and mental functioning19,20(the Dutch translation of the SF-36 was validated in both the general population and populations with chronic disease21); the Hospital Anxiety and Depression Scale (HADS) for assessment of self-reported anxiety and depression22,23; the Dissociation Experience Scale for measurement of self-reported dissociative experiences24; and the Neuropsychiatric Inventory (NPI), a 10−20 min interview, for evaluation of a wide range of NP symptoms, recording severity and frequency separately25. Psychopathology is based on the psychiatric history and mental status examination, following Diagnostic and Statistical Manual IV classification26.
Neuropsychology
Formal neuropsychological testing of patients, including history taking and clinical observation, is conducted to obtain quantitative measures of global cognitive functioning with a specific focus on memory, executive functioning, and psychomotor speed as adapted from the neuropsychological test battery suggested by the 1999 ACR NPSLE nomenclature and case definition system, Appendix C5. Global cognitive functioning of patients is assessed using the Mini Mental State Examination. The Wechsler Memory Scale and the Wechsler Adult Intelligence Scale-Revised (WAIS-R) subtest Digit Span are used to examine memory functions. Executive functions are assessed with the Stroop Color and Word Test, the Trail Making Test, the WAIS-R subtest Digit Symbol Coding, a Word Fluency Task, and the Digit Cancellation Test. The Digit Symbol Coding test also provides a measure of psychomotor speed. Patients’ handwritten copies of perspective and geometric figures are used as measures for constructional praxis. Cognitive performance also depends on the psychiatric status of the patient. Therefore, the HADS and NPI are also part of the neuropsychological examination. Details regarding administration, scoring, and clinical value of the neuropsychological tests have been described27. If indicated, patients are scheduled for a second session for additional neuropsychological testing. The neuropsychological examination is evaluated by an experienced clinical neuropsychologist (HAMM). Cognitive deficits are classified as definitive, questionable, or absent as interpreted by the clinical neuropsychologist. Cognitive deficits are considered severe if they result in inability to function in daily life without professional help.
Radiology
Standard of imaging is magnetic resonance imaging (MRI), performed on a 3 Tesla MRI scanner (Philips Medical Systems), and images are evaluated by an experienced neuroradiologist (MvB)28. The scanning protocol consists of a high-resolution T1-weighted sequence before and after intravenous administration of gadolinium contrast agent, T2-weighted, fluid-attenuated inversion recovery (FLAIR) sequences, and a diffusion-weighted imaging (DWI) sequence. In addition to the standard clinical sequences, diffusion tensor images (DTI), magnetization transfer imaging (MTI), proton magnetic resonance spectroscopic imaging, and resting state functional MRI are performed. Previous studies have shown that MTI is a valuable addition in diagnosing NPSLE, by correlating change in peak height to clinical activity of the disease29. Infarction on MRI is defined as tissue loss or parenchymal defect, following the signal intensities of CSF (i.e., high on T2 and low on T1 and FLAIR) with a surrounding area of high signal on T2 and FLAIR respecting the flow territories and not explained by trauma or iatrogenic lesions30. If indicated, MR angiography of cerebral arteries and veins or MRI of the spine is performed.
Laboratory tests
Laboratory evaluation is performed, including a complete blood count, creatinine clearance, urinalysis, liver function tests, electrolytes, erythrocyte sedimentation rate and C-reactive protein, anti-dsDNA antibodies, rheumatoid factor, antinuclear factor, antiextractable nuclear antigens, complement levels, thyroid function, lipid profile, and glucose. Anticardiolipin antibodies (aCL) and lupus anticoagulant are measured once. A serum sample and DNA from all patients is stored for future research purposes.
Consensus meeting
All medical specialists above meet in 2 weekly scheduled meetings to discuss the patients. Diagnosis of NPSLE is made by consensus, taking into account the assessments described above. The consensus group agrees upon the following aspects: (1) diagnosis of SLE15,16; (2) objective complaints (assessed to standard of care of the appropriate medical specialty); (3) absence of another diagnosis that explains symptoms (e.g., schizophrenia in psychosis); (4) diagnosis of NPSLE and ACR 1999 classification5 if appropriate; and (5) suspected pathogenetic mechanism for NPSLE and advice on therapy. To diagnose and classify patients, all the assessments are considered; Figure 1 offers an algorithm of the most relevant diagnostic considerations based on the expert opinion of the consensus group. A representative of every discipline is required for the consensus meeting and after discussion, conclusions were unanimous.
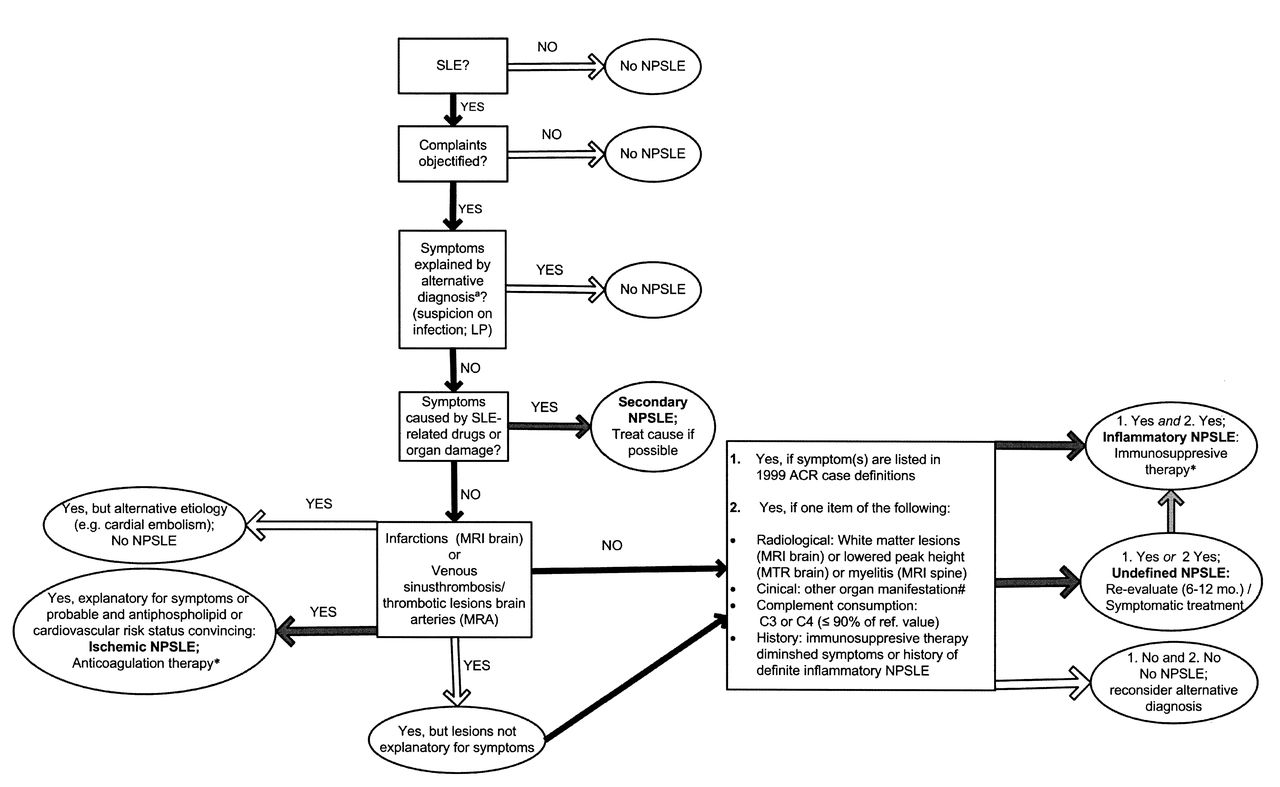
The algorithm of the most relevant diagnostic considerations based on the expert opinion of the consensus group. LP: lumbar puncture; MRI: magnetic resonance imaging; MRA: magnetic resonance angiography; MTR: magnetization transfer ratio. aIncluding all exclusion criteria of the American College of Rheumatology (ACR) 1999 neuropsychiatric systemic lupus erythematosus (NPSLE) case definitions; other items considered: family history, psychosocial conditions, age, and timing of symptom onset. *Treat all cardiovascular risk factors. #Indication for therapy regardless of NPSLE status, including hematological manifestations.
For each patient we assess involvement of the following pathogenetic mechanisms: (1) primary inflammatory NPSLE (inflammatory and neurotoxic pathways); (2) primary ischemic NPSLE (ischemic and thrombotic pathways); (3) undefined NPSLE; and (4) secondary NPSLE (NP symptoms secondary to medication for SLE or organ damage related to SLE). A patient with inflammatory or ischemic NPSLE will be advised to be treated with the respective immunosuppressive or anticoagulation therapy, following international recommendations31. Anticoagulants can be added to immunosuppressive therapy in patients with inflammatory disease with signs of secondary ischemia. Patients with undefined NPSLE will be advised to be treated symptomatically only (anticonvulsants, antidepressants, antipsychotics, or psychological therapy), or are reevaluated in 6 to 12 months if symptoms persist. In patients with secondary NPSLE, advice focuses on the specific cause, and current medication is evaluated and change advised if appropriate.
Further, the following descriptors are noted: chronology (episodic, remittent, sustained, progressive), severity [mild (patient is able to lead a normal daily life), moderate, severe (prolonged hospital stay due to inabilities from neurological/psychiatric disorder, death)], following the ACR 1999 criteria for basic descriptors5. With respect to immunosuppressive medication, the consensus group agrees on the following: patients with severe symptoms according to ACR 1999 criteria5 get the advice for cyclophosphamide intravenously (National Institutes of Health regime) for at least 6 months, in accord with 3 consecutive days of methylprednisolone 1000 mg intravenously, followed by 1 mg/kg prednisolone orally tapered with 10 mg/month. Patients with mild symptoms are advised 0.5 mg/kg prednisolone orally and patients with moderate symptoms are advised 1 mg/kg prednisolone orally. When a prolonged course of symptoms is expected we advise azathioprine to be added to oral prednisolone for maintenance therapy and taper prednisolone as soon as possible.
Data analysis
Descriptive statistics are used for the patients’ characteristics. Comparisons between phenotypes are done with Mann-Whitney U test and chi-squared tests where appropriate. Characteristics of patients of the phenotype under study are compared with characteristics of all patients with SLE of other phenotypes. All significant results are reported. Sensitivity and specificity are calculated for all dichotomous variables that show significance.
RESULTS
From September 2007 until December 2009 we evaluated 100 patients. The feasibility of the NPSLE clinic was evaluated with respect to necessity and organization. The assessment program took place every week as scheduled. We were able to conduct all assessments as outlined in Table 1 in 97 patients. In 2 patients, NP examination was not conducted because of coma and severe symptoms of psychosis, and in 1 patient MRI of the brain was not performed because of claustrophobia. Of the 100 evaluated patients, 70 and 13 were referred to the NPSLE clinic by a rheumatologist or neurologist, respectively. Seventeen patients were referred by other medical specialists, e.g., an internist or psychiatrist. More than 60% of patients were referred by a medical specialist from other hospitals, of which 15% worked in a university medical center.
Figure 2 shows the outcome of the evaluation of NP symptoms and the advice for therapy in the first 100 patients that were evaluated. Seventy-one patients fulfilled ACR criteria for SLE and 46 were diagnosed with NPSLE. Twenty-nine patients did not fulfill ≥ 4 ACR criteria15,16. If the algorithm in Figure 1 is considered, 97 out of the 100 evaluated patients were correctly classified and received advice that was consistent with the expected phenotype. Two incorrectly classified patients received advice for symptomatic treatment and therefore are described as undefined NPSLE; however, considering the algorithm they should have been classified as inflammatory NPSLE. The third misclassified patient received advice for antiplatelet medication and therefore was classified as ischemic NPSLE; however, following the algorithm this patient should have been classified as undefined NPSLE.

Outcome of the evaluation of neuropsychiatric (NP) symptoms and the advice for therapy in the first 100 patients evaluated. Seventy-one patients fulfilled American College of Rheumatology criteria for systemic lupus erythematosus (SLE) and 46 were diagnosed with neuropsychiatric SLE (NPSLE). *Combined with corticosteroids. MMF: mycophenolate mofetil.
Thirty-eight patients (38/71; 54% of all patients with SLE) were diagnosed with primary NPSLE. Figure 2 shows the advice for therapy for all patients. Twenty-one patients (21/38; 55%) were diagnosed with inflammatory NPSLE and were advised to be treated with immunosuppressive medication. One patient with inflammatory NPSLE was treated with mycophenolate mofetil because of coexisting renal involvement, and in 1 patient with inflammatory NPSLE, antiplatelet therapy was added to immunosuppressive medication. Twelve patients (12/38; 32%) were diagnosed with ischemic NPSLE and were advised treatment with anticoagulant medication. In 2 of these patients antiplatelet therapy was added to current oral anticoagulants. Five (5/38; 13%) patients were diagnosed with undefined NPSLE.
Eight patients (8/71; 11% of all patients with SLE) were diagnosed with secondary NPSLE. Of them, 7 (7/8; 88%) were advised to change their medication, which meant reducing corticosteroids in 5 patients (5/8; 63%). One patient had cognitive dysfunction because of irreversible damage caused by SLE 20 years before evaluation.
Twenty-five patients with SLE (25/71; 35%) had NP symptoms that were not attributed to SLE. In 3 patients (3/25; 12%) the NP symptoms (cognitive dysfunction in 2 patients and intermittent loss of consciousness in 1 patient) could not be objectified. In 22 patients (22/25; 88%) NP symptoms were attributable to other causes, in the majority to preexisting psychiatric syndromes like schizophrenia or depression or to psychosocial circumstances. In this category we also found NP symptoms due to multiple sclerosis, cervical disc herniation, and an arachnoidal cyst in the brain.
Table 2 shows sociodemographic and clinical characteristics of patients by clinical phenotype. Definitive cognitive dysfunction was more prevalent in inflammatory NPSLE compared to SLE patients of other phenotypes (p < 0.005; sensitivity and specificity of cognitive dysfunction for diagnosis of inflammatory NPSLE 62% and 74%, respectively). Further, in inflammatory NPSLE, disease activity was relatively high (mean SLEDAI 9.7, SD 5.4), due partly to the presence of the NP symptoms (mean SLEDAI excluding NP symptoms was 5.2, SD 2.8). Disease activity in patients with inflammatory NPSLE was significantly higher than that in patients with SLE of other phenotypes (SLEDAI p < 0.005; SLEDAI excluding NP symptoms p < 0.05). Further, in inflammatory NPSLE, 16 patients (16/21; 76%) had moderate and 4 had severe symptoms (4/21; 19%). Ten patients (10/21; 48%) showed a chronic disease course, 4 (4/21; 19%) a progressive course, and 4 (4/21; 19%) either an episodic or a remittent course.
Sociodemographic and clinical characteristics of 71 SLE patients with neuropsychiatric manifestations.
In ischemic NPSLE, IgG aCL were highly prevalent; this was significantly different from patients of other clinical phenotypes (p < 0.05; sensitivity and specificity of IgG aCL for diagnosis of ischemic NPSLE 58% and 81%, respectively). Prevalence of other CV risk factors noted in Table 2 did not differ significantly between patients with ischemic NPSLE and patients of other phenotypes. In contrast to other phenotypes, only patients with ischemic NPSLE reported a TIA (3/12; 25%). MRI abnormalities are classification requirements and therefore are present in 100% of patients with ischemic NPSLE, and although MR imaging is also frequently abnormal in other phenotypes, MRI presents a distinguishable characteristic for ischemic NPSLE (p < 0.05; sensitivity 100% and specificity 40%). In patients with ischemic NPSLE, severity of symptoms was considered to be moderate in 92% (11/12). Half of these patients had 1 disease episode, whereas the others showed either a remittent or a chronic disease course.
History of renal disease is highly prevalent in patients with secondary NPSLE, compared with patients of other phenotypes, a significant difference (p < 0.05; sensitivity and specificity of renal disorders for the diagnosis of secondary NPSLE 50% and 84%, respectively). Moreover, patients with secondary NPSLE used significantly more corticosteroids than patients of other phenotypes (p < 0.05; sensitivity and specificity of prescription of corticosteroids for diagnosis of secondary NPSLE 88% and 57%). Symptoms were mild in all patients with secondary NPSLE, with 3 patients (3/8; 38%) having a chronic disease course, 2 (2/8; 25%) a remittent, and 3 (3/8; 38%) an episodic course.
Patients with undefined NPSLE did not differ significantly from patients of other phenotypes. Most patients had an episodic disease course and moderate symptom severity.
Table 3 shows diagnoses according to ACR case definitions of patients with primary NPSLE5. In undefined and inflammatory NPSLE, 2 and 4 patients, respectively, had 1 ACR 1999 diagnosis, and 2 and 10 patients had 2 diagnoses. In inflammatory NPSLE, 7 patients had 3 diagnoses. In ischemic NPSLE, 1 patient had 1 diagnosis, 6 had 2 diagnoses, 4 had 3 diagnoses, and 1 patient had 4 diagnoses.
ACR 1999 NPSLE diagnoses in primary NPSLE patients. Patients can have multiple diagnosis. ACR 1999 diagnoses not found in our patients include aseptic meningitis, demyelinating syndrome, acute confusional state, acute inflammatory demyelinating polyradiculopathy, autonomic disorder, myasthenia gravis, cranial neuropathy, and plexopathy. Data are number (%).
Fourteen patients (14/21; 67%) with inflammatory NPSLE had an ACR diagnosis of cognitive dysfunction; in 2 (2/21; 10%) cognitive dysfunction was severe. Of the 2 other patients with severe cognitive dysfunction, 1 had ischemic NPSLE, and 1 non-NPSLE patient was diagnosed with Alzheimer’s disease.
DISCUSSION
To our knowledge this is the first prospective evaluation of a standardized, multidisciplinary assessment of NP symptoms in patients with SLE. Of our patients, half were diagnosed with primary NPSLE. This level of attribution of NP symptoms directly to SLE is in agreement with data from a Canadian cohort, although that study lacked a standard multidisciplinary assessment32. In primary NPSLE, most patients experience inflammatory NPSLE followed in frequency by ischemic NPSLE, and in a small proportion, undefined NPSLE.
Inflammatory NPSLE is best characterized by high disease activity and cognitive dysfunction. Although a considerable part of the SLEDAI is based on NP symptoms, SLEDAI results with exclusion of NP symptoms are still significantly higher in this phenotype. Disease activity is a known risk factor for NPSLE33,34.
High prevalence of cognitive dysfunction in NPSLE is in accord with recent studies3,4. The recent advances in experimental science, linking autoantibodies to cognitive dysfunction, underline the relevance of cognitive dysfunction in inflammatory NPSLE1,2. However, attribution of cognitive dysfunction to SLE is arguable in many patients35. In our cohort we also encountered cognitive dysfunction in non-NPSLE patients, but prevalence of cognitive dysfunction in inflammatory NPSLE is distinctly different. In contrast, prevalence of mood disorders and headache was surprisingly similar in all groups, even in non-NPSLE. The lack of specificity of headache as a symptom of NPSLE is underscored by results from a case-control study and a meta-analysis36,37. With respect to mood disorders, this is reinforced by evidence that depression and anxiety in patients with SLE are related to psychosocial circumstances, intrusiveness of illness, and symptom concealment rather than to SLE-specific variables38,39. Hence cognitive dysfunction is a specific feature of inflammatory NPSLE, in contrast to symptoms such as headache or mood disorders.
Ischemic NPSLE is best characterized by high prevalence of IgG aCL and the presence of abnormalities on MRI. In patients with ischemic NPSLE, traditional CV risk factors (diabetes, hypertension, obesity, smoking, and total cholesterol) are not markedly increased. Traditional risk factors are known to be inadequate to explain ischemic events and its precursors in patients with SLE40,41. In contrast, IgG aCL did aid in diagnosing ischemic NPSLE in our cohort. NP manifestations in relation to the antiphospholipid syndrome, whether primary or secondary, are described in the literature42,43,44.
In our expert opinion, based on the algorithm, abnormalities on MRI present a key item for classification of a patient into the ischemic NPSLE phenotype. Therefore a sensitivity of 100% for MRI in ischemic NPSLE is expected because of the circular reasoning of the judgment of MRI findings; what is surprising, however, is the poor specificity (40%), which is due to abnormalities on MRI in other phenotypes of NPSLE and ischemic lesions that were not explanatory for the current symptoms. TIA were reported in the group of patients with ischemic NPSLE only; pathogenetically this is to be expected, however, the findings could be of value in clinical practice. To question patients with SLE on TIA is a simple, safe, and inexpensive procedure and proved to be relevant as it could be specifically indicative of ischemic NPSLE.
Secondary NPSLE is best characterized by a history of renal disease and by the use of corticosteroids. Corticosteroids are a potential cause of NP symptoms, by contrast this mechanism should be excluded in primary NPSLE5. The high prevalence of history of renal disease in this phenotype could be reflective of overall, including cerebral, organ damage or could be the result of transient electrolyte disturbances.
Although clinical assessments show more abnormalities in patients with NPSLE, non-NPSLE patients do not have exclusively normal results. This emphasizes the lack of a “gold standard” for NPSLE and the presence of nonspecific NP manifestations in patients with SLE35. A diagnosis based on multidisciplinary consensus after a standardized assessment is currently the best strategy for diagnosing and classifying NPSLE and is therefore an appropriate reference standard45.
The described clinical phenotypes show similarities that probably partly represent the true picture of clinical characteristics in patients of different phenotypes. Nonetheless, when more patients have been assessed, different phenotypes of NPSLE could become more distinguishable. The selection of our patient cohort likely was biased through referral by their treating physicians. However, since it is plausible that patients with a clear diagnosis will be referred less often, the diagnostic value of specific characteristics is likely underestimated in our cohort.
Our study also shows the feasibility and necessity of a dedicated clinic for NPSLE. In the future, we will investigate the accuracy of our diagnoses and the effect of therapy started in the cohort described here, reevaluating the patients in a multidisciplinary followup visit. Eventually, instead of diagnoses and classification of NPSLE based on expert opinion, we aim to establish a validated model that can be used for therapeutic decisions in NPSLE. Therapeutic strategies based on clinical phenotypes could become even more important when advances in experimental science lead to new targets for therapy in NPSLE46.
We have described a multidisciplinary diagnostic approach for NPSLE and its clinical phenotypes based on the suspected pathogenetic mechanism. Three defined NPSLE phenotypes show some remarkable features, although characteristics also overlap. The characteristics found to be most helpful in the diagnostic process are disease activity and cognitive dysfunction with respect to inflammatory NPSLE, and abnormalities on MRI and IgG aCL for ischemic NPSLE. Secondary NPSLE is best characterized by the use of corticosteroids, often also the cause for NP symptoms.
- Accepted for publication July 17, 2012.








{kind=link}
{kind=link}