

Abstract
In this literature review, the mechanisms underlying pain associated with osteoarthritis (OA) are discussed, along with evidence for the efficacy of medications thought to act centrally to relieve OA pain. We survey the cascade of events from inflammation to activation of nociceptive and neuropathic pathways, to the development and maintenance of central and peripheral sensitization. Preclinical and clinical evidence for the sensitization hypothesis is discussed, along with recently identified genetic variations that may increase sensitivity to pain in patients with OA. Evidence is presented for the efficacy of centrally acting analgesics for OA pain (opioids, antiepileptics, tricyclic antidepressants, and serotonin/norepinephrine receptor inhibitors).
Osteoarthritis (OA) represents a heterogenous group of painful conditions involving a common pathophysiology at the affected joint1. The pain of OA is believed to be driven by both nociceptive and neuropathic mechanisms as well as abnormal excitability in the pain pathways of the peripheral and central nervous system2. Pain is the most prominent symptom of OA, and is strongly associated with reduced functional ability3. Loss of function and pain are weakly correlated to severity of disease by radiography4. Outside the articular cartilage, joint structures are richly innervated and are potential sources of pain in OA5. However, there is often discordance between the degree of articular pathology and pain experienced by patients with OA, suggesting the presence of individual variability in the central processing of nociceptive stimuli. Several studies have demonstrated the presence of hyperalgesia6,7,8, abnormal pain sensitivity7,9,10, and referred pain11,12,13 in patients with OA. Despite the disabling role of pain from OA, much remains to be clarified about its pathophysiology. Existing pharmacologic interventions for pain from OA are frequently insufficient or poorly tolerated; consequently, pain from OA is often inadequately controlled14. Greater appreciation for the pain pathways involved, including central sensitization, may have significant implications for a more comprehensive framework of analgesic therapy for patients with OA.
We conducted a literature review regarding the pathophysiology of OA pain.
Pathophysiology of pain
Nociceptive pain arises from stimulation of peripheral nerve endings. The nociceptive signal is transmitted through the spinal cord to the brain along multiple neuronal pathways15. Descending pathways from the brain to the spinal cord modulate pain signals, thereby increasing or decreasing pain perception16. Activation of central nociceptive pathways (e.g., dysregulation of neurotransmission or damage to the central nervous system) without involving peripheral nociceptors can also generate pain17. Additionally, inflammatory mediators released in damaged tissue and in the spinal cord can sensitize the peripheral terminals of nociceptors or the spinal neurons of the ascending nociceptive pathways15. Changes in peripheral and central neurons result in lower pain thresholds, secondary hyperalgesia in uninjured areas, and sustained pain potentiation.
Although a key pathologic hallmark of OA is abnormal articular cartilage, cartilage is avascular and hence aneural. Therefore, damaged articular cartilage is incapable of directly generating the pain of OA18. Most studies indicate only half of patients with radiographic OA have pain, and there is a weak correlation of pain severity to radiographic features19. Hence, some patients with minimal cartilage abnormality have significant pain, and some patients with severe cartilage abnormalities have minimal or no pain.
Peripheral mechanisms
Multiple nociceptors may be involved in transmitting pain signals in OA. Thinly myelinated Aδ (group III) and unmyelinated C (group IV) fibers innervate the synovial capsule, periarticular ligaments, menisci, adjacent periosteum, and subchondral bone. The thicker myelinated, proprioceptive Aß (group II) fibers terminate in the synovial capsule, periarticular bursae, fat pad, ligaments, menisci, and adjacent bony periosteum2,20. Aß fibers are generally activated by normal joint movement. The Aδ and C fibers are activated by noxious mechanical, thermal, or chemical stimuli. Additionally, in severe OA, blood vessels breach the articular cartilage tidemark. This vascularization leads to sensory [i.e., substance P and the calcitonin-gene-related peptide (CGRP)] and sympathetic nerve (i.e., C-flanking peptide of neuropeptide) invasion of articular cartilage in patients with OA21.
Inflammation in the joint triggers a cascade of events, resulting in peripheral sensitization, increased sensitivity of nociceptive primary afferent neurons, and hyperexcitability of nociceptive neurons in the central nervous system22. As the cartilage is destroyed, chondrocytes produce inflammatory mediators and degradative enzymes, resulting in eventual cellular apoptosis23. This cascade of events is initiated by the inflammatory stimuli and driven by cytokines including tumor necrosis factor-α and various proinflammatory interleukins, chemokines, nerve growth factor, sympathetic amines, leukotrienes, prostaglandins, and matrix metalloproteinases2,24. Inflammatory mediators, such as bradykinin, histamine, lactic acid, substance P, and CGRP, induce hyperalgesia by a number of direct and indirect actions, triggering a self-propagating cycle in which more inflammatory mediators are released18,24. Macrophage-mediated inflammation can directly and indirectly stimulate angiogenesis by secreting factors to stimulate endothelial cells and fibroblasts to produce angiogenic factors such as vascular endothelial growth factor25. Additionally, hypoxia in inflamed tissue is a potent stimulator of angiogenesis, causing further inflammation and pain through the formation of new vessels and the innervation of these vessels. Endochondral ossification and the formation of osteophytes occur at the osteochondral junction in response to angiogenesis, further exacerbating the inflammation and associated pain.
Bone-related causes of pain in OA include subchondral microfractures, bone stretching with elevation of periosteum due to osteophyte growth, bone remodeling and repair, bone marrow lesions, and bone angina caused by decreased blood flow and elevated intraosseous pressure25,26. Bone marrow lesions (also called bone edema) detected on magnetic resonance imaging (MRI) appear to reflect bone infarction, and are associated with higher levels of OA pain and a greater likelihood of disease progression26.
While pain associated with OA has long been considered nociceptive pain coupled with local inflammatory mechanisms, there is a growing hypothesis that neuropathic mechanisms may also be contributing to the pain experience in some patients with OA27. The molecular mechanisms possibly contributing to a neuropathic-like pain in OA are poorly described. However, because many of the structures in the joint are richly innervated, it is possible that the local damage to these and other joint structures could cause damage to the peripheral nerves20.
Although the molecular mechanisms are poorly characterized, there is some evidence that patients with OA may experience neuropathic-like pain. Focus groups of patients with OA of the knee or hip gave detailed descriptions of the pain associated with their OA conditions28. Some patients used words such as burning and numbness to describe the pain associated with OA; these descriptors suggest a neuropathic pain component.
Central sensitization in OA
Central sensitization is defined as a state in which neurons activated by nociceptive stimuli are sensitized by such stimuli and become hyperresponsive to subsequent stimuli to the neuron’s receptor fields29. Peripheral sensitization is thought to play an important role in the development and maintenance of central sensitization. Intense, repeated or prolonged input from peripheral nociceptors modulates spinal cord pain-transmitting neurons and leads to decreased activation thresholds, increased synaptic excitability, and increased firing thresholds30,31.
A growing body of evidence reveals a significant contribution of excess nociceptive ascending and deficient inhibitory descending influences in the development and maintenance of central sensitization. The signals are carried from the dorsal horn along the ascending pain pathways to the brain stem, hypothalamus, and thalamus, and the cerebral cortex, where pain is interpreted (Figure 1)32,33. The lateral system and the medial system of the lateral spinothalamic tract are responsible for the perception of pain34. Descending pathways modulate activity in the dorsal horn by controlling spinal pain transmission16,33. As a result, central nociceptor transmission neurons in the dorsal horn of the spinal cord can be regulated.
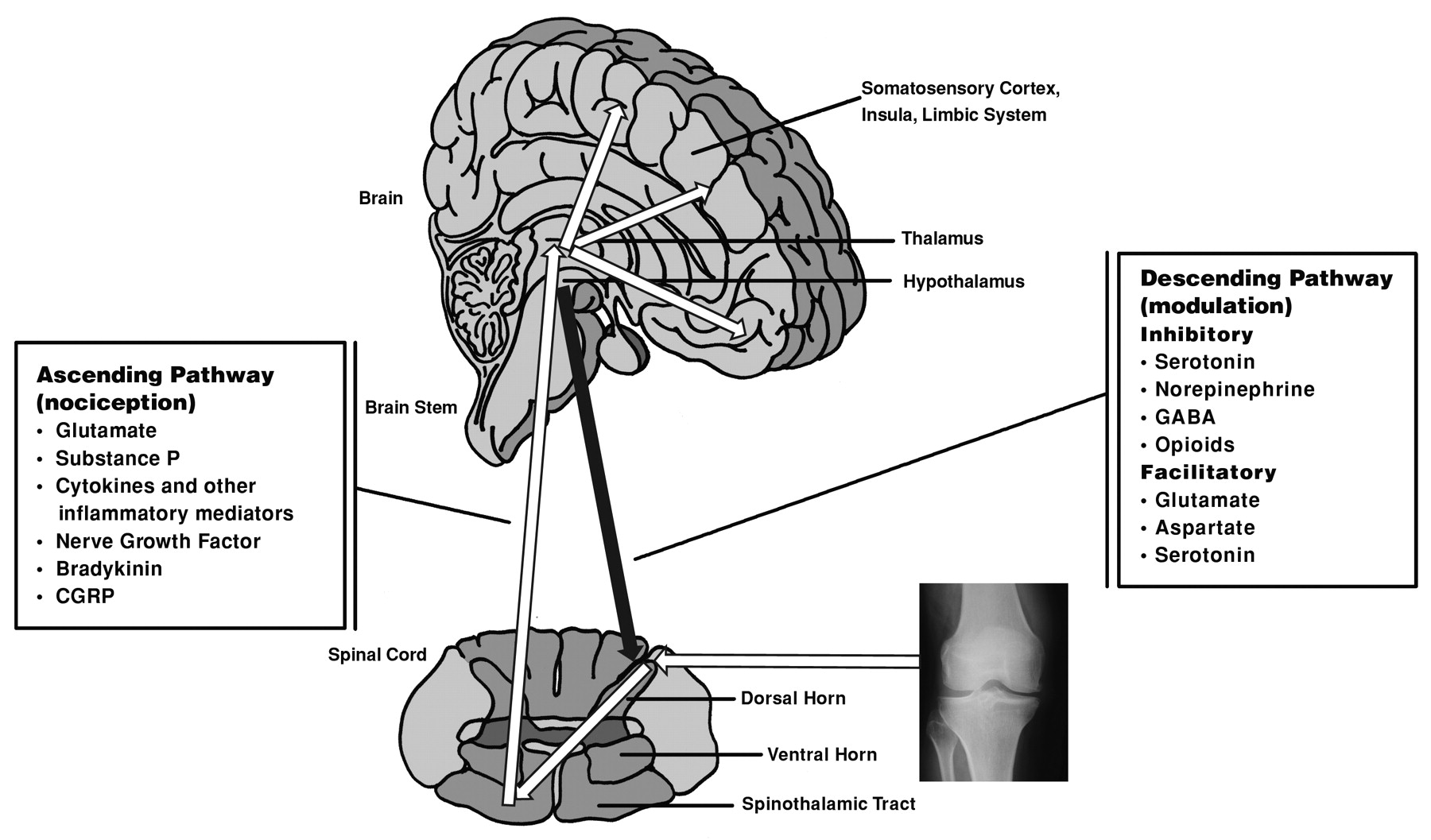
Neurologic pain pathways and associated neurotransmitters thought to be active in pain perception associated with osteoarthritis. Ascending pathways are nociceptive. Descending pathways modulate the response to painful stimuli at the level of the dorsal horn. Modulating fibers can either facilitate or inhibit pain signaling through the ascending pathway. (Illustration is modified from the original, provided courtesy of Patrick J. Lynch, medical illustrator; C. Carl Jaffe, MD, cardiologist.) http://commons.wikimedia.org/wiki/File:Skull_and_sagittal_brain.svg?uselang=fr.html#globalusage.
Central sensitization contributes to the temporal, spatial and threshold changes and the generation of pain hypersensitivity experienced in acute and chronic pain settings29. Both preclinical and clinical evidence support the hypothesis that central sensitization may be involved in the processing of OA pain. Findings of intraspinal release of prostaglandin E2 accompanied by an upregulation of the cyclooxygenase-2 enzyme in rat models of acute arthritis of the knee suggest that intraspinal prostaglandins play a role in inflammation-evoked central sensitization of spinal cord neurons35. Additionally, the hyperalgesia described in experimental arthritis models in animals has been thought to represent dorsal horn mechanisms36,37,38. Study of thermal and mechanical pain thresholds in patients with OA of the hands demonstrated that central mechanisms contribute to hyperalgesia related to movement pain6,39.
Persistent nociceptive input from an osteoarthritic joint is believed to result in central sensitization. To further test the theory of central sensitization in OA, infusion of hypertonic saline into the lower extremities was used to determine whether patients with OA would report increased muscle hyperalgesia and enlarged referred and radiating pain areas compared with healthy control subjects11. Infusion of hypertonic saline in the tibialis anterior muscle evoked muscle pain with a longer duration and intensity in patients with OA, and larger pain areas with significantly increased referred and radiating pain intensity than in control subjects, suggesting central sensitization. Intraarticular local anesthetic may have both peripheral and central effects on pain in OA. In patients with bilateral symptomatic OA of the knee, injection of intraarticular anesthetic in 1 knee produced decreases in pain perception in both knees40. Decreased pain in the contralateral knee cannot be attributed to the systemic effect of the anesthetic due to rapid dilution; therefore, the reduced pain in the contralateral knee is likely to be due to decreased activity of a spinal mechanism driven from peripheral input. In a majority of patients with OA of the knee who have failed to respond adequately to conservative nonpharmacological therapy and simple analgesics, intraarticular depocorticosteroids or intraarticular hyaluronan can reduce pain41,42.
Despite small sample sizes, evidence suggests activation of descending inhibitory pathways, in addition to peripheral and central sensitization to stimuli in patients with OA. Thresholds to cutaneous mechanical sensation (both innocuous and abnormal noxious stimulation) and pain were analyzed in both knee joints of patients with either rheumatoid arthritis or OA as well as in both knee joints of control subjects7. Simultaneous mechanical allodynia and cutaneous hypoesthesia adjacent to the affected joint, in addition to paradoxical decreased sensation and increased pain thresholds to mechanical stimulation, suggest that central mechanisms may be involved in the pain associated with OA.
During a series of pain sensitivity studies in patients with knee OA, phasic pressure stimuli were applied to 4 sites bilaterally in the patients’ shoulders and arms43. Significantly higher levels of pain intensity were reported in the patients with OA than in healthy control subjects. No difference between the 2 groups was observed in response to thermal heat. These results are consistent with the hypothesis that central sensitization and perhaps spinal glial cell activation contribute to pain sensitivity and modulation in OA. One study investigated the supraspinal influences that underlie referred pain and changes in skin sensitivity in patients with OA awaiting joint replacement compared with age-matched control subjects using pain psychology questionnaires, quantitative sensory testing, and functional MRI44. Patients with OA had significantly greater activation in the brainstem (periaqueductal gray matter) than healthy control subjects, as illustrated on functional MRI in response to punctate stimulation of their referred pain areas. Additionally, the magnitude of this activation positively correlated with the extent of neuropathic-like elements of the pain described by the patient. These findings suggest a central target is attributable for analgesia and that neuropathic mechanisms may be involved in OA pain. Despite the evidence, however, many questions remain unanswered regarding the contribution of central sensitization and neuropathic mechanisms to OA pain.
Role of genetics in OA pain
Evidence suggests that genetic variations, such as low catechol-O-methyltransferase (COMT) enzyme (encoded by the COMT gene; 22q11) activity, may increase sensitivity to pain45,46. Indeed, the low-activity allele polymorphism (Val158Met) of COMT was associated with OA pain in women with both radiographic hip OA and symptoms of hip OA47. If replicated, the results of this study may have important clinical implications and may help explain the differences in symptom severity in patients with identical pathologic findings.
Treatment of osteoarthritis
The primary goal of OA treatment is directed at controlling pain, improving function, and improving a patient’s quality of life. Treatment guidelines by the American College of Rheumatology for chronic OA are currently under review and not yet published. Treatment of OA should be tailored to each individual patient based on clinical assessment, as recommended by the Osteoarthritis Research Society International. Our review focuses on the role of centrally acting analgesics in OA, such as opioids, antiepileptics, tricyclic antidepressants (TCA), and serotonin/norepinephrine receptor inhibitors (SNRI). The role of peripherally acting analgesics such as nonsteroidal antiinflammatory drugs (NSAID) is beyond the scope of our review. While these centrally acting agents have different mechanisms of action and are approved for other indications, they are often used to treat that part of OA pain that originates from central rather than peripheral mechanisms and thus may complement the peripherally acting analgesic and antiinflammatory medications.
Weak opioids and narcotic analgesics with or without acetaminophen are useful alternatives in patients for whom NSAID are contraindicated, or for whom combined acetaminophen, opioids, and NSAID therapy is ineffective42. Stronger opioids have a role in a limited number of patients with severe pain. Certain opioids have been reported to be efficacious for the management of OA pain48,49,50, such as those for which therapeutic action is mainly attributed to the activation of μ-opioid receptors and to the inhibition of serotonin and norepinephrine reuptake mechanisms, as well as various voltage-gated and ligand-gated ion channels of the nociceptive system.
Antiepileptics are also used to treat chronic pain conditions. Analgesic efficacy has been demonstrated for agents having a high binding affinity to the α2-Δ site (an auxiliary subunit of voltage-gated calcium channels) in central nervous system tissues51,52. The mechanism may be related to reduced calcium-dependent release of several neurotransmitters, such as glutamate and substance P. Evidence in animal models suggests that antiepileptics may be potential analgesic targets in pain associated with OA53,54. Further research is required to determine the effects of antiepileptics during the treatment of pain in patients with OA.
Medications originally developed as antidepressants, such as TCA and SNRI, are used to treat some chronic pain conditions55,56,57. The analgesic effects of these agents appear to be independent of their antidepressant effects58. TCA and SNRI are thought to relieve pain by blockade of serotonin and norepinephrine reuptake into presynaptic neurons, thus augmenting the effect of these neurotransmitters on important inhibitory pain pathways. The TCA may also inhibit NMDA receptors and inhibit ion channel activity56. SNRI are sometimes used with other medications and may provide an alternative to patients who cannot tolerate or have a contraindication for other forms of therapy. SNRI appear to act at least partially by augmenting the function of serotonin and norepinephrine in modulation of noxious pain signaling59. Evidence is increasing regarding the analgesic role of SNRI. Significantly reduced pain associated with arthritis was reported in elderly patients with comorbid major depressive disorder and arthritis treated with an SNRI60. In a single-blind, placebo run-in study, treatment with an SNRI significantly reduced pain intensity (as measured by the Western Ontario and McMaster Universities Osteoarthritis Index pain scale) in patients with OA61. In 2 randomized, double-blind, placebo-controlled trials, patients with OA of the knee treated with an SNRI had a significantly greater reduction in pain compared with placebo, and also demonstrated improvement in function62,63.
OA is characterized by degenerative and inflammatory processes affecting joints and surrounding tissues, which are clinically manifested by pain and functional disability. Pain manifestations in OA are thought to be driven by joint pathophysiology and abnormal excitability in peripheral and central pain pathways. Although there are a limited number of clinical studies demonstrating the role of central sensitization in pain associated with OA, evidence suggests that central mechanisms are at least partially responsible for the pain of OA, and there is preliminary evidence that suggests the potential benefit of centrally acting medicines to treat OA pain. Additionally, increasing evidence supports the hypothesis that neuropathic mechanisms may also be contributing to the pain associated with OA and patients may benefit from neuropathic pain medications. Further research is warranted to examine the roles of central mechanisms and neuropathic pain in OA. Greater understanding of the potential mechanisms involved in the pathophysiology can lead to a more targeted treatment approach.
Footnotes
-
Supported by Eli Lilly and Company.
- Accepted for publication March 16, 2011.








{kind=link}
{kind=link}