

To the Editor:
A 71-year-old woman presented with a 7-day history of a tender rash and necrosis on both shins. Six months prior to admission, she first noticed intermittent occurrence of purpura on her legs. She had been taking non-steroidal antiinflammatory drugs (NSAID) for her joint pain for 2 months and was not taking any anticoagulants. She was a nonsmoker with no history of miscarriages or allergies. Her history was unremarkable and no family history of autoimmune diseases, cancer, diabetes, or coagulopathies was elicited.
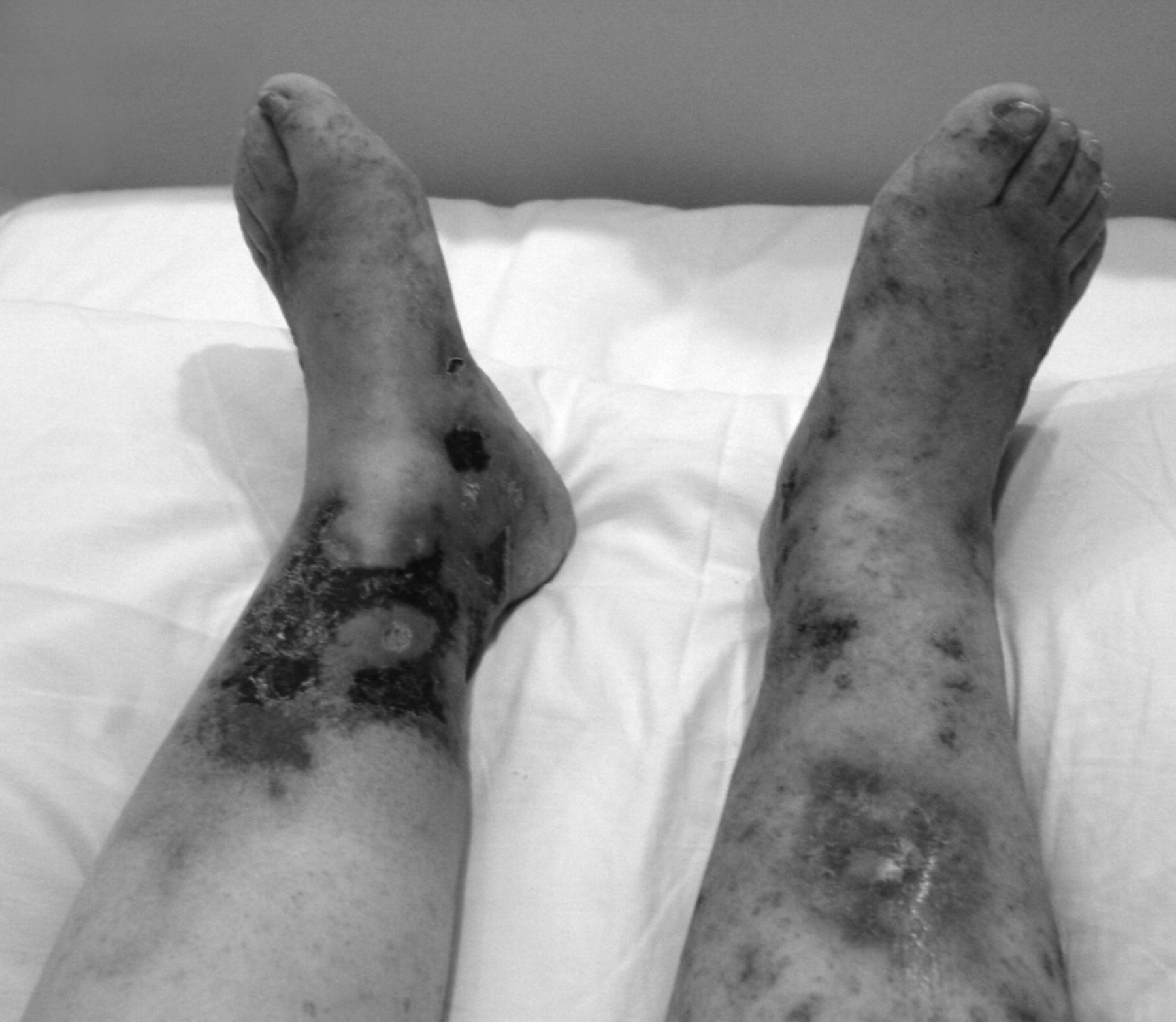
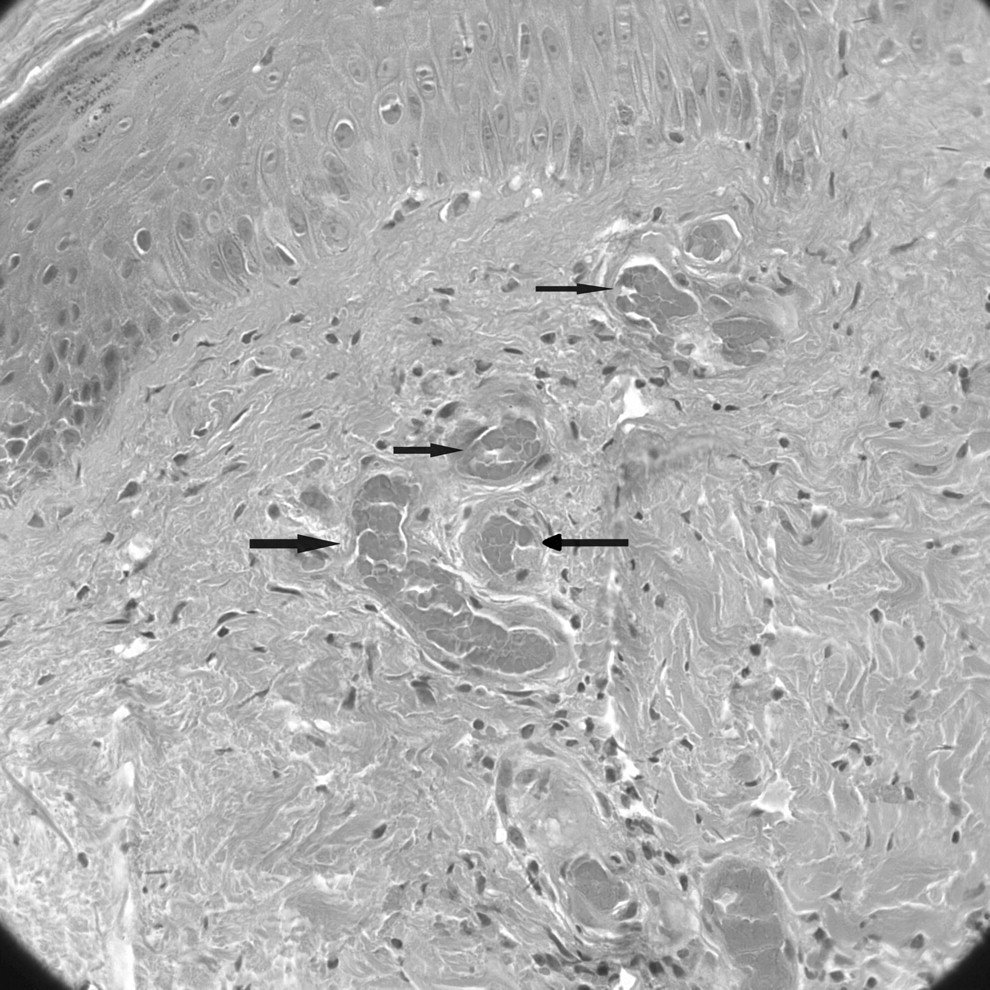
On examination, she was afebrile with no cardiovascular, respiratory, or abdominal findings. On the lower limbs, there was bilateral involvement with skin necrosis on the medial distal third, more pronounced on the left, which was surrounded by purpura and ecchymosis. In addition, there were erythema and purpura, more confluent on the dorsum of the feet, but also scattered on both lower limbs (Figure 1). Routine investigations including full blood count, urea and electrolytes, and coagulation were normal. The erythrocyte sedimentation rate was elevated, 72 mm/h (normal range 0–20), but antinuclear antibody was within normal limits. Venous ultra-sonography of both legs showed no abnormalities, but the arterial Doppler scan showed several atheromatous plaques in the arteries of her lower limbs. Subsequently, magnetic resonance imaging of the brain revealed multiple small ischemic lesions in both frontal lobes and lateral ventricles. Histopathology demonstrated thrombi inside the small vessels and extravasated red blood cells in the superficial dermis. There was little inflammatory infiltration and no fibrinoid degeneration of vascular walls (Figure 2).
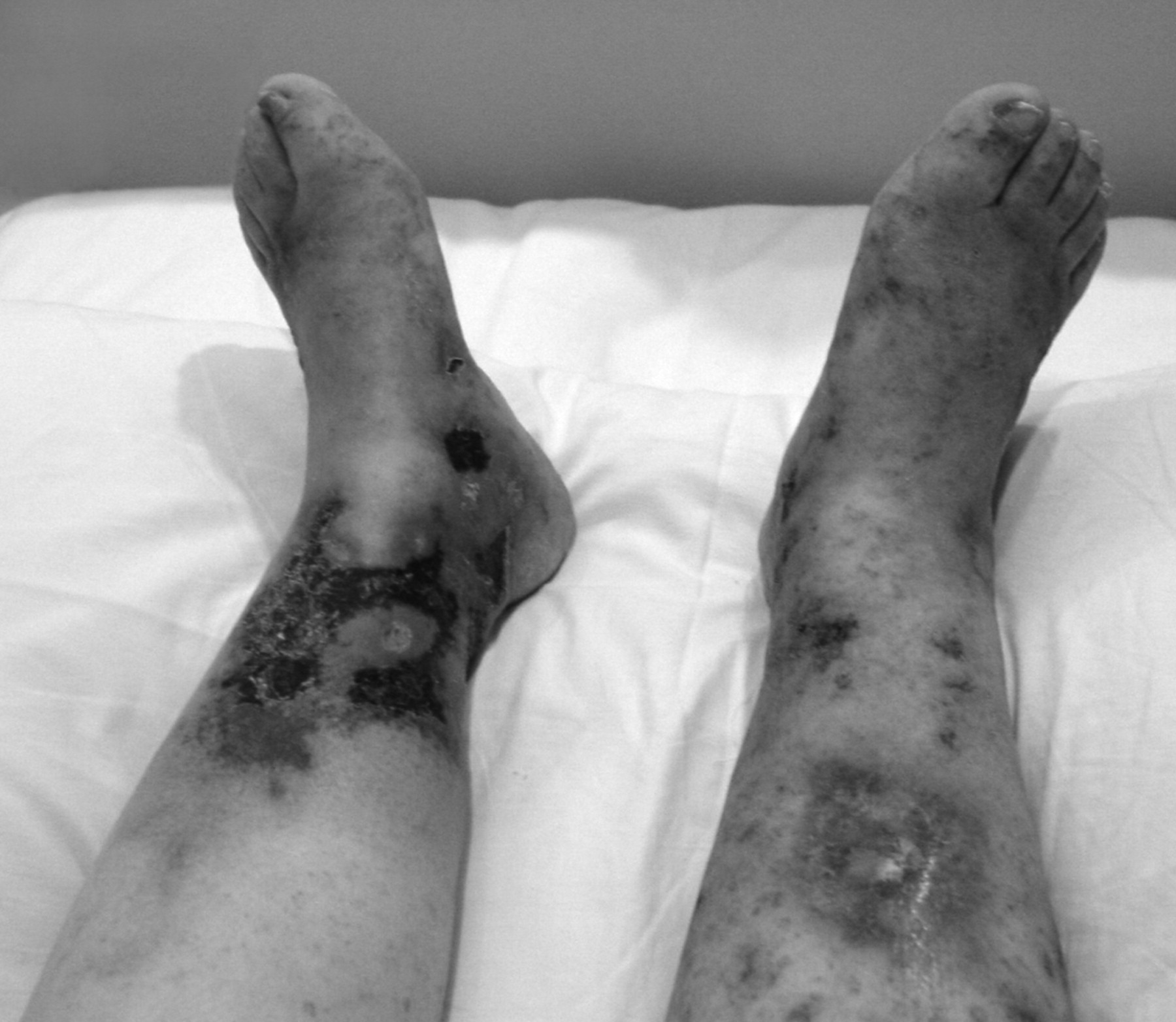
Cutaneous necrosis in lower limbs.
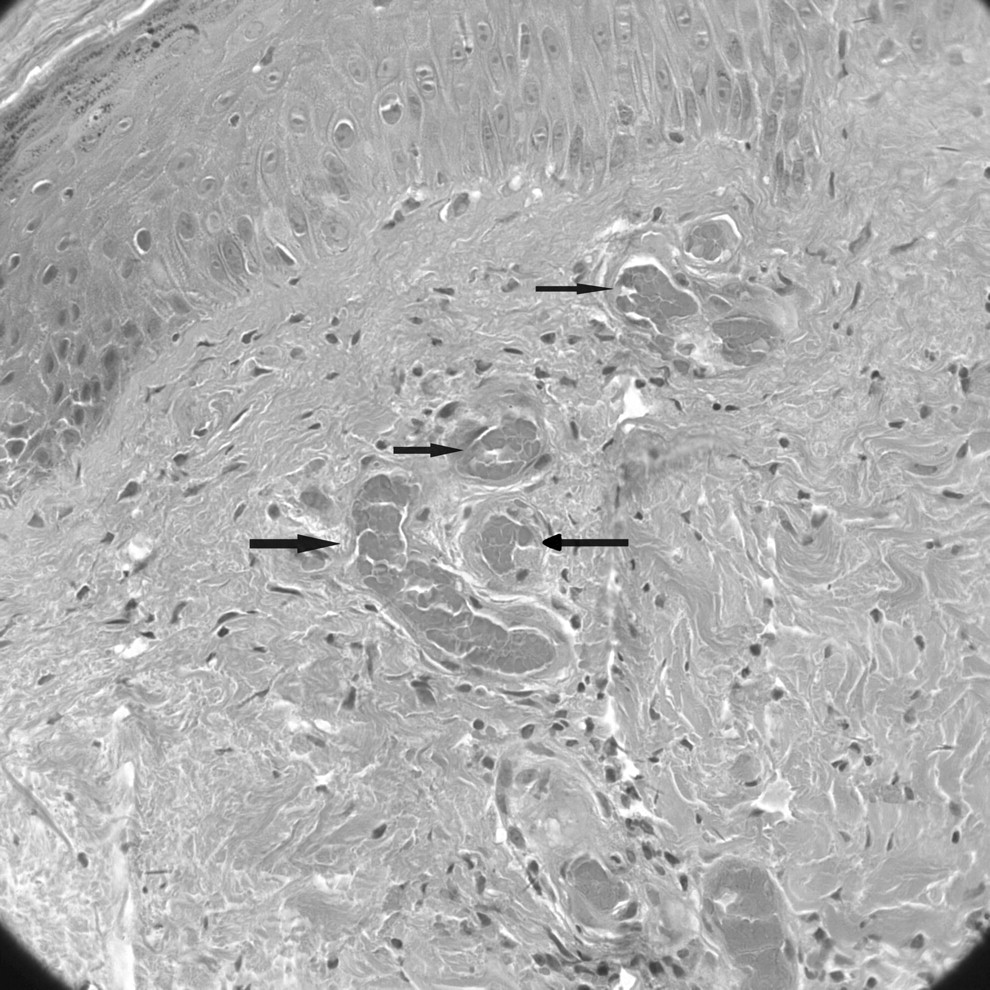
Skin biopsy from the left lower leg showing thrombi inside the small vessels (H&E stain, original magnification ×20).
The following diagnoses should be considered. (1) Systemic necrotizing vasculitis: Lesions can be limited to the skin, with a predominant distribution in the extremities, but is diagnosed by fibrinoid degeneration and/or vascular wall necrosis on histopathology (absent in this case). (2) Henoch-Schönlein purpura (HSP): Despite the occurrence of joint pain with purpura in this patient, HSP is unlikely as an exceedingly painful necrosis is rarely seen in patients with HSP. In addition, HSP histopathology usually reveals leukocytoclastic vasculitis. (3) Purpura fulminans (also known as “purpura gangrenosa”): This diagnosis should be considered when there is a sudden appearance of symmetrical, tender, ecchymotic skin lesions. Characterized by cutaneous hemorrhage, necrosis, and disseminated intravascular coagulopathy, purpura fulminans is much more common in children and is usually associated with severe sepsis. Our patient was in the elderly age group and was not septic.
Other differentials include necrotizing fasciitis, deep vein thrombosis, pyoderma gangrenosum, erysipelas, and cutaneous necrotizing venulitis associated with connective tissue disease.
Further investigations revealed raised anticardiolipin IgM, 48.0 (normal < 12.0 MPL/ml); positive lupus anticoagulant and anti-ß2 glycoprotein-I antibody and a normal anticardiolipin IgG of 7.9 (normal < 12.0 GPL/ml). The patient was diagnosed with primary antiphospholipid syndrome (APS) and subsequently started lifelong anticoagulation therapy, initially with low molecular weight heparin and aspirin, which was replaced with warfarin when international normalized ratio (INR) levels became therapeutic and stabilized between 2 and 3. The skin lesions resolved rapidly after 3 weeks of treatment and at 3 months, anticardiolipin IgM was still elevated, 23.0 MPL/ml, while IgG was in the normal range; lupus anticoagulant was weakly positive. At 6 months, she remained asymptomatic.
First described in 1983, APS is a multisystem autoimmune disease, characterized by persistent raised titers of antiphospholipid antibodies with one or more of the following: recurrent arterial or venous thrombosis or recurrent miscarriages1. APS often presents in young women of childbearing age, with only 12.7% of patients presenting after 50 years of age2,3. Our case, where the patient developed APS in her seventies with cutaneous presentation only, is extremely rare.
Clinical manifestations of APS are diverse and may involve any of the respiratory, cardiovascular, cerebral, renal, dermatological, and hematological systems4. As described by Cervera, et al2, primary APS, which refers to APS occurring in isolation, as opposed to secondary APS, which is associated with other autoimmune diseases, occurred in 53.1% in a cohort of 1000 patients with APS. Dermatological findings, although not pathognomonic, are important in the diagnosis of APS. Cutaneous necrosis, one of the recognized features in patients with APS, usually presents acutely with painful purpura5,6,7. Common sites involved are the extremities, cheek, trunk, forehead, and helices of ears8. Other skin findings include livedo reticularis (most frequently associated with cutaneous APS manifestation), leg ulcers, pseudovasculitis, digital gangrene, and splinter hemorrhages5,6.
APS is an important differential to consider in unexplained lower limb necrosis. Once this diagnosis is achieved, it is astute to start patients at once on anticoagulation rather than antiinflammatory therapy.








{kind=link}
{kind=link}