

To the Editor:
Juvenile dermatomyositis (JDM) is an immune-mediated pediatric idiopathic inflammatory myopathy (IIM) characterized by muscle and skin inflammation1. Anti-PL-12 is a myositis-specific antibody (MSA), directed against alanine-tRNA synthetase2, often with debilitating interstitial lung disease (ILD), that is sporadically reported in childhood3. We describe a child with anti-PL-12 antibody who recovered lung function after therapy.
A 14-year-old African American girl developed bilateral ankle stiffness associated with swelling and redness of the upper eyelid. One month later, she noticed proximal weakness of shoulder girdle and pelvic girdle. Four months later, she had fever to 102°F; an erythematous rash over upper chest, arms, and upper eyelids; difficulty walking and getting up from bed, with shortness of breath during exertion. After admission to intensive care unit for suspected aspiration pneumonia, she was diagnosed with JDM on the basis of a muscle biopsy. She received intravenous (IV) steroid (30 mg/kg/dose) for 5 days, followed by oral prednisone (1 mg/kg/day), and was lost to followup.
Six months later, she was admitted to Children’s Memorial Hospital with fever, malaise, weakness, worsening rash, and bilateral knee arthritis. Laboratory data were elevated: creatine kinase 12,464 IU/l (normal 29–165 IU/l), aldolase 150 U/l (normal 3.4–8.6 U/l), alanine transaminase 156 IU/l (normal 2–30 IU/l), aspartate transaminase 257 IU/l (normal 16–52 IU/l), and lactate dehydrogenase 1496 IU/l (normal 126–289 IU/l). Markers of inflammation were increased: erythrocyte sedimentation rate (ESR) 37 mm/h (normal < 20 mm/h), C-reactive protein (CRP) 4.41 mg/dl (normal < 0.8 mg/dl), von Willebrand factor antigen (vWF:Ag) 420% (blood group B normal = 57%–241%); and elevated neopterin level 30.4 nm/l (normal < 10 nm/l). Periungual capillaroscopy showed dilated capillaries, and moderately severe dropout of the nailfold capillary end-row loop (ERL): 4.28 (normal value 7–10)4.
Tests for anti-dsDNA, Scl-70, and rheumatoid factor were negative, but her sera were positive for anti-Ro and anti-PL-12 (laboratory of Dr. I. Targoff5). On pulmonary function testing, she had decreased forced vital capacity, 73%, and diffusion capacity (DLCO/VA) of 66%, consistent with restrictive lung disease (Table 1). Computed tomography (CT) scan of the chest showed peripheral intralobular and interlobular septal thickening, early parenchymal cystic changes, and micronodules consistent with ILD (Figure 1). Dual energy X-ray absorptiometry scan showed a lumbar Z score of −1.9 (consistent with osteopenia) and vitamin D deficiency (25-hydroxy D total 25.2 ng/ml; normal 30–119 ng/ml]. Over the next 17 months, she developed persistent osteopenia (lumbar Z score of −2.6), despite supplementation with calcium and vitamin D and attaining a therapeutic level of vitamin D.

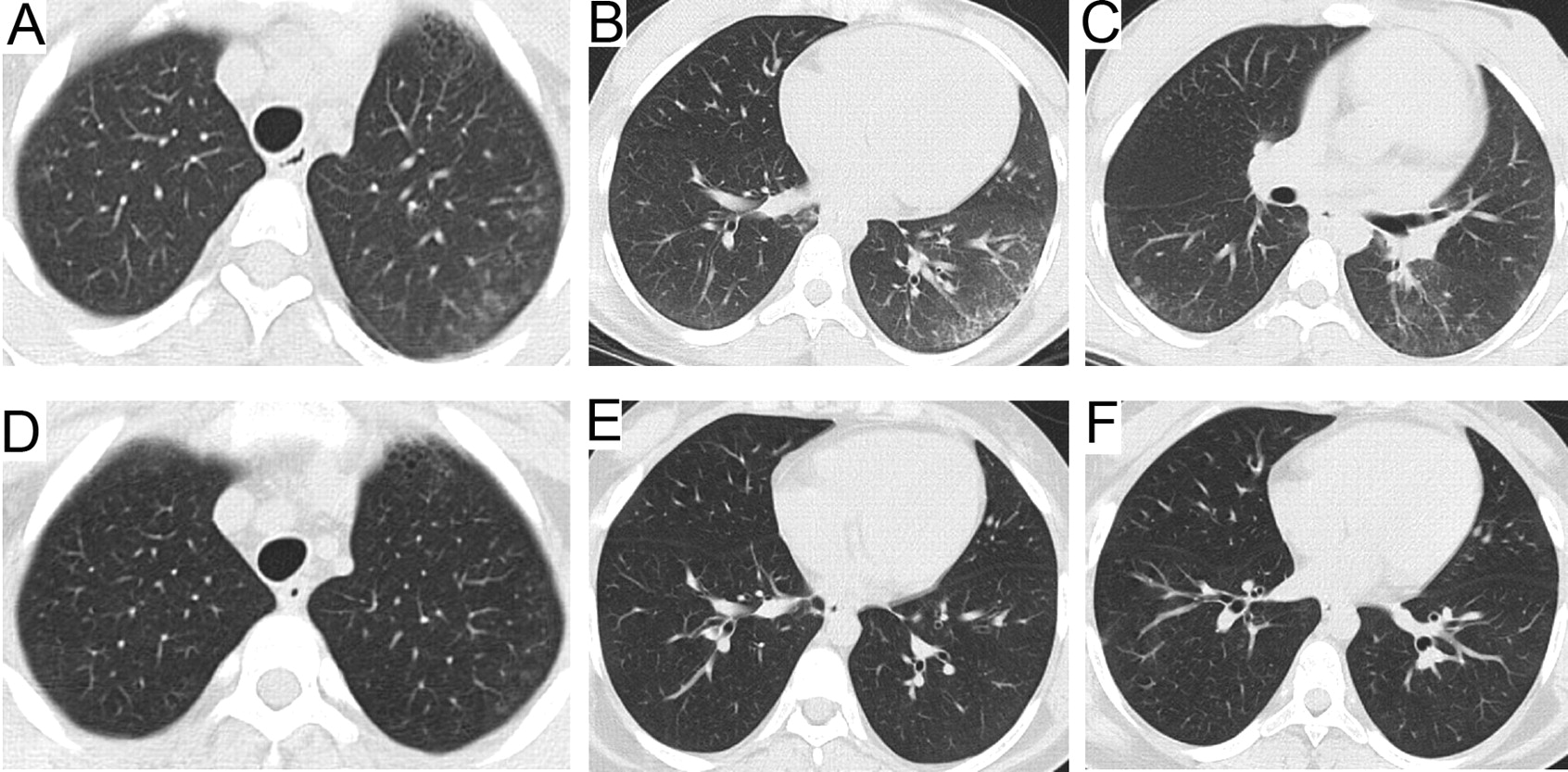
CT scans before treatment showing peripheral intralobular interstitial thickening at the lung bases, left greater than right (A, B, C); and after treatment showing resolution of previous changes (D, E, F).
Lung function data over time (dates shown). All data are percentages.
Therapy included 1 g IV methylprednisolone (IVMP), on 3 consecutive days, followed by twice weekly for the first 3 months; oral prednisolone 20 mg/day (0.43 mg/kg) on non-pulse days; methotrexate IV followed by subcutaneous (SC; 15 mg/m2) weekly and monthly IV cyclophosphamide (500 mg/m2) for 6 months. One month after completion of cyclophosphamide she had a normal leukocyte count, and she was given mycophenolate mofetil (MMF) 1000 mg daily (21 mg/kg/day) and continued with IVMP pulses every other week for 8 months and SC methotrexate weekly. IV administration of methylprednisolone was utilized because of prolonged active vasculitis characterized by a persistent rash associated with microvascular damage reflected by loss of nailfold capillary ERL, as well as positive indicators of immune activation and endothelial cell damage (elevated levels of vWF:Ag, low C4, decreased CD3-negative natural killer cells)6. Rouster-Stevens, et al showed that patients with JDM and ERL loss may have decreased bioavailability of oral prednisone compared with IVMP7. IVMP was tapered only after symptoms and all laboratory data were normal, including muscle enzymes and other markers of inflammation (ESR, CRP, and vWF:Ag), following the authors’ treatment protocol for severe JDM8. Two years after starting therapy at Children’s Memorial Hospital, she takes prednisone 5 mg daily (0.1 mg/kg/day); SC methotrexate 15 mg/m2 every week; MMF 2500 mg daily (40 mg/kg/daily); folic acid 1 mg; calcium 1600 mg; and vitamin D 50,000 units/month. The pulmonary function essentially normalized after 5 months of therapy and the serologic indices of inflammation normalized after 17 months. Her CT scan showed resolution of interstitial and interlobular septal thickening with minimal residual micronodules (Figure 1).
The most common myositis-specific antibodies are directed against aminoacyl-tRNA synthetase, one of the enzymes catalyzing the attachment of a particular amino acid to its transfer RNA2. Patients with aminoacyltRNA synthetase have the anti-synthetase syndrome: myositis, polyarthritis, fever, Raynaud’s phenomenon, ILD, and mechanic’s hands9. In a study of 77 children, 12 (16%) had myositis-specific antibodies and one had anti-PL-123. Myositis-specific antibodies are found in 16%–26% of adults with IIM2; anti-PL-12 is present in < 5%. Anti-PL-12 is highly associated with ILD5; in 31 adults, ILD was present in 90%, and preceded the diagnosis of connective tissue disease in 53%10.
Our patient’s pulmonary function tests showed a pattern of restrictive lung disease, commonly associated with diminished core strength as well as a diffusion defect11. The chest CT identified interlobular septal thickening, one of the most common radiological findings in PL-12-positive adults. Although myositis is usually mild in patients with anti-PL-125,10, our patient had severe myositis with markedly elevated muscle enzymes, and her score on the Childhood Myositis Assessment Scale12 was markedly decreased at 21/52. She was negative for antibody to ribonuclear protein, which is present in 36%–65% of patients who are anti-PL-12-positive10. VWF:Ag is often an indicator of a disease flare in IIM13,14, and our patient exhibited high vWF:Ag levels at presentation, persisting for 9 months. Her disease course was similar to those of adults with both anti-PL-12 and anti-Ro antibody, in whom ILD is difficult to control. In contrast to reported cases, however, her pulmonary function tests normalized and the chest CT scan showed significant resolution of ILD. To our knowledge this is the first complete report of a child with IIM with both anti-PL-12 and anti-Ro autoantibodies and improvement of lung function in response to intensive immunosuppressive therapy.
Footnotes
-
Supported by NIH/NIAMS R01 AR48289, the Cure JM Foundation, and the Macy’s Miracle Foundation.








{kind=link}