

Abstract
Objective. Inhibition of intracellular signal transduction is considered to be a therapeutic target for chronic inflammation. The new Janus kinase (JAK)3 inhibitor CP690,550 has shown efficacy in the treatment of rheumatoid arthritis (RA). We investigated the influence of JAK/STAT inhibition using CP690,550 on the induction of acute-phase serum amyloid A (SAA), which is triggered by interleukin 6 (IL-6) stimulation in rheumatoid fibroblast-like synoviocytes (RA-FLS).
Methods. IL-6-stimulated gene expression of the acute-phase serum amyloid A genes (A-SAA; encoded by SAA1+SAA2) and SAA4 was analyzed by reverse transcriptase-polymerase chain reaction. The intracellular signaling pathway mediating the effects of CP690,550 on IL-6-stimulated JAK/STAT activation was assessed by measuring the phosphorylation levels using Western blots.
Results. IL-6 trans-signaling induced A-SAA messenger RNA (mRNA) expression in RA-FLS. By contrast IL-6 stimulation did not affect SAA4 mRNA expression, which is expressed constitutively in RA-FLS. IL-6 stimulation elicited rapid phosphorylation of JAK2 and STAT3, which was blunted by CP690,550. CP690,550 abrogated IL-6-mediated A-SAA mRNA expression in RA-FLS. Similarly, CP690,550 inhibited IL-6-mediated A-SAA mRNA expression in human hepatocytes.
Conclusion. Our data indicated that CP690,550 blocked IL-6-induced JAK2/STAT3 activation, as well as the induction of A-SAA. Inhibition of IL-6-mediated proinflammatory signaling pathways by CP690,550 may represent a new antiinflammatory therapeutic strategy for RA and AA amyloidosis.
- AA AMYLOIDOSIS
- CP690,550
- SERUM AMYLOID A
- RHEUMATOID ARTHRITIS
- SIGNAL TRANSDUCERS AND ACTIVATORS OF TRANSCRIPTION
- JANUS KINASE
Rheumatoid arthritis (RA) is characterized by synovial inflammation and subsequent cartilage damage1. The varying levels of inflammation in RA are reflected by alterations in the acute-phase response, which is characterized by increased serum acute-phase protein2. Serum amyloid A (SAA), one of the acute-phase proteins, is the precursor of amyloid A protein, the fibrillar component of AA amyloidosis3. SAA is regulated by inflammatory cytokines, and abundant levels of SAA have been demonstrated in rheumatoid synovium4,5. Further, SAA plays a central role in the proinflammatory cascade of RA, since SAA exhibits a number of proinflammatory properties6. For example, SAA induces migration of inflammatory cells and the expression of inflammatory cytokines, including interleukin 6 (IL-6)7,8. Moreover, SAA can stimulate the production of cartilage-degrading proteinases, such as matrix metalloproteinases (MMP)9.
In humans, 4 SAA genes have been described. Two genes (SAA1 and SAA2) encode A-SAA and are concurrently induced in response to inflammation10. A-SAA increases dramatically during acute inflammation and may reach levels that are 1000-fold greater than normal10. A-SAA is mainly synthesized in the liver, but extrahepatic production has been described11. SAA is produced in response to proinflammatory cytokines, such as IL-1, IL-6, and tumor necrosis factor-α (TNF-α)12. Since proinflammatory cytokines are markedly elevated in patients with RA, anticytokine therapy has been introduced13. Among these treatments, IL-6-blocking therapy has shown promise by normalizing serum levels of SAA in patients with RA14. Moreover, it has been demonstrated that blocking IL-6 alone, but not IL-1 or TNF-α, completely blocked A-SAA messenger RNA (mRNA) expression in human hepatocytes during triple cytokine stimulation with IL-6, IL-1, and TNF-α15. For signal transduction, IL-6 binds to the membrane-bound IL-6 receptor (gp80)16. The IL-6-gp80 dimer then interacts with gp130. Formation of gp130-containing complexes results in the activation of Janus kinases (JAK), which activates signal transducers and activators of transcription (STAT)17. Recent evidence suggests that STAT3 is the key transcription factor responsible for IL-6 activation of A-SAA gene transcription18. CP690,550 is an orally available JAK antagonist that is currently in development for the treatment of RA19,20. Although the drug was initially thought to be a JAK3-specific inhibitor, recent studies raise the possibility that some of the efficacy of CP690,550 might to be due to inhibition of JAK1 or JAK2 as well as JAK321. The aim of our study was to identify the role of JAK inhibition using CP690,550 in the IL-6 signaling pathway that leads to the induction of A-SAA in rheumatoid synovium.
MATERIALS AND METHODS
Reagents
The JAK inhibitor CP690,550 was obtained from Axon Biochemicals (Groningen, The Netherlands). AG490 was obtained from Calbiochem (San Diego, CA, USA). Human recombinant IL-6 and soluble IL-6 receptor (sIL-6R) were purchased from Peprotech (Rocky Hills, NJ, USA). Phospho-specific and pan antibodies against JAK-1 (Tyr1022/1023), JAK-2 (Tyr1007/1008), STAT-1 (Tyr701), STAT-3 (Tyr705), and STAT-5 (Tyr694) were purchased from Cell Signaling Technology (Beverly, MA, USA). Phospho-specific and pan antibodies against JAK3 (Tyr980) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Immunohistochemistry
For immunohistochemical analysis for SAA, formalin-fixed and paraffin-embedded tissue blocks were cut into sections 4 μm thick. Sections were deparaffinized in xylene and subsequently rehydrated in sequential ethanol (100%–70%). After washing 3 times with 10 mmol/l phosphate-buffered saline (PBS; pH 7.4), antigen retrieval was performed by first heating in a microwave at 95°C for 20 min, then by washing twice in PBS for 10 min. The sections were treated with peroxidase-blocking solution (Dako Japan, Kyoto, Japan) for 5 min, and incubated with 1:1000 dilution of rabbit anti-SAA polyclonal antibody22. A standardized 2-step method with Envision Plus (Dako) was used for detection. Reaction products were visualized using diaminobenzidine as a chromogen (Dako), and counterstained with Mayer’s hematoxylin (Dako).
Preparation of fibroblast-like synoviocytes (FLS)
Synovial tissue was obtained from patients with RA at the time of total joint replacement or synovectomy. All patients with RA fulfilled the American College of Rheumatology criteria23. Synovium was minced and incubated with 1 mg/ml collagenase type VIII (Sigma-Aldrich, St. Louis, MO, USA) in serum-free RPMI-1640 medium (Life Technologies, Grand Island, NY, USA) for 1 h at 37°C, filtered, washed extensively, and cultured in Dulbecco modified Eagle’s medium (Life Technologies) supplemented with 10% fetal bovine serum in a humidified 5% CO2 atmosphere. FLS were used from passages 4 through 7, when they are a homogeneous population (< 1% CD45-positive).
Human primary hepatocytes were purchased from Cell Systems (Kirkland, WA, USA). The cells were cultured in a basal medium composed of Ham’s F-12 and Leibovitz L-15 (1:1) medium (Invitrogen, Carlsbad, CA, USA), 0.2% (vol/vol) bovine serum albumin, 5 mM glucose (Wako Chemical, Osaka, Japan), 10−8 M dexamethasone (Wako), and 10−8 M bovine insulin (Invitrogen) supplemented with 10% (vol/vol) fetal calf serum (Gibco, Grand Island, NY, USA).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from FLS using the RNeasy total RNA isolation protocol (Qiagen, Crawley, UK). Total cellular RNA was extracted with Trizol (Invitrogen) according to the manufacturer’s protocol. First-strand cDNA was synthesized from 1 μg of total cellular RNA using an RNA PCR kit (Takara Bio Inc., Otsu, Japan) with random primers. Thereafter, cDNA was amplified using specific primers, as follows. For A-SAA: forward primer 5′-CGA AGC TTC TTT TCG TTC CTT-3′; reverse primer 5′-CAG GCC AGC AGG TCG GAA GTG-3′. SAA1: forward primer 5′-CGA AGC TTC TTT TCG TTC CTT-3′; reverse primer 5′-CAC CAT GGC CAA AGA ATC TC -3′. SAA2: forward primer 5′-ATG GGG CTC GGG ACA TGT GGA G-3′; reverse primer 5′-AGT CCT CCG CAC CAT GGC CAA A-3′. SAA4: forward primer 5′-CCA GTG AAA GCT GGC GTT CG-3′; reverse primer 5′-GAG AAG TGT GTG GCT CAC AGC C-3′24. C-reactive protein (CRP): forward primer 5′-ACT TCC TAT GTA TCC CTC AAA G-3′; reverse primer 5′-CTC ATT GTC TTG TCT CTT GGT-3′. ß-actin forward primer: 5′-GTG GGG CGC CCC AGG CAC CA-3′; reverse primer 5′-CTC CTT AAT GTC ACG CAC GAT TTC-3′.
The product sizes were 300 bp for A-SAA, 217 bp for SAA1, 192 bp for SAA2, 138 bp for CRP, and 234 bp for ß-actin. Thermocycling conditions (35 cycles) for the targets were as follows: A-SAA and SAA4: 94°C for 60 s and 53°C for 60 s, and 72°C for 60 s. SAA1: 95°C for 60 s and 59°C for 60 s, and 72°C for 60 s. SAA2: 94°C for 60 s and 59°C for 60 s, and 72°C for 60 s. CRP and ß-actin: 94°C for 60 s and 56°C for 60 s, and 72°C for 60 s.
PCR products were electrophoresed on 2% agarose gels and visualized by ethidium bromide staining. The density of A-SAA, SAA1, SAA2, and ß-actin bands was determined by densitometer (NIH Image system; National Institutes of Health, Bethesda, MD, USA) and the ratio of each band against ß-actin was calculated.
Cell lysis and Western blotting
Serum-starved RA-FLS were stimulated with IL-6 and sIL-6R for the times indicated in the figure legends, and the cells were washed with ice-cold PBS and lysed with a lysis buffer (1% Nonidet P40, 50 mM Tris, pH 7.5, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 20 mM ß-glycerophosphate, 1.0 mM sodium orthovanadate, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) for 20 min at 4°C. Insoluble material was removed by centrifugation at 15,000 g for 15 min at 4°C. The supernatant was saved and the protein concentration was determined using the Bio-Rad protein assay kit (Bio Rad, Hercules, CA, USA). An identical amount of protein (50 μg) for each lysate was subjected to 10% SDS-polyacrylamide gel electrophoresis, and then transferred to a nitrocellulose membrane. Western blot analysis using phospho-specific anti-JAK and STAT antibodies was performed with an ECL Western blotting kit (Amersham, Little Chalfont, UK).
RESULTS
Serum amyloid A expression in rheumatoid synovium
We examined whether RA-FLS produce SAA using immunohistochemical techniques. Positive SAA staining was observed in synovial fibroblasts in addition to vascular endothelial cells and their lumens (Figure 1A). SAA protein was not detected in osteoarthritis synovial tissue samples (Figure 1B).

SAA protein expression in rheumatoid synovial tissues. A. Tissue sections stained using antibodies specific for SAA protein. SAA was localized in synovial fibroblasts in addition to the endothelial cells (arrows). A representative result of 3 independent experiments (original magnification ×200). B. Synovial tissue sections from a patient with osteoarthritis (OA) were stained using antibodies specific for SAA protein. SAA localization was not confirmed in OA synovial tissues. A representative result of 2 independent experiments (original magnification ×200).
Acute-phase serum amyloid A mRNA expression in IL-6-stimulated RA-FLS
RT-PCR analysis was used to determine A-SAA mRNA expression in RA-FLS. A-SAA mRNA was not detected in RA-FLS under control conditions. However, A-SAA mRNA expression was abundantly induced when RA-FLS were stimulated with IL-6 plus sIL-6R, but not when RA-FLS were stimulated with IL-6 or sIL-6R alone (Figure 2A). We also analyzed SAA1 and SAA2 mRNA in RA-FLS using RT-PCR analysis and confirmed that expression of SAA1 and SAA2 mRNA was induced by IL-6 plus sIL-6R stimulation (Figure 2B, 2C). SAA4 mRNA was constitutively expressed in RA-FLS in the presence or absence of IL-6 or sIL-6R stimulation (Figure 2D).
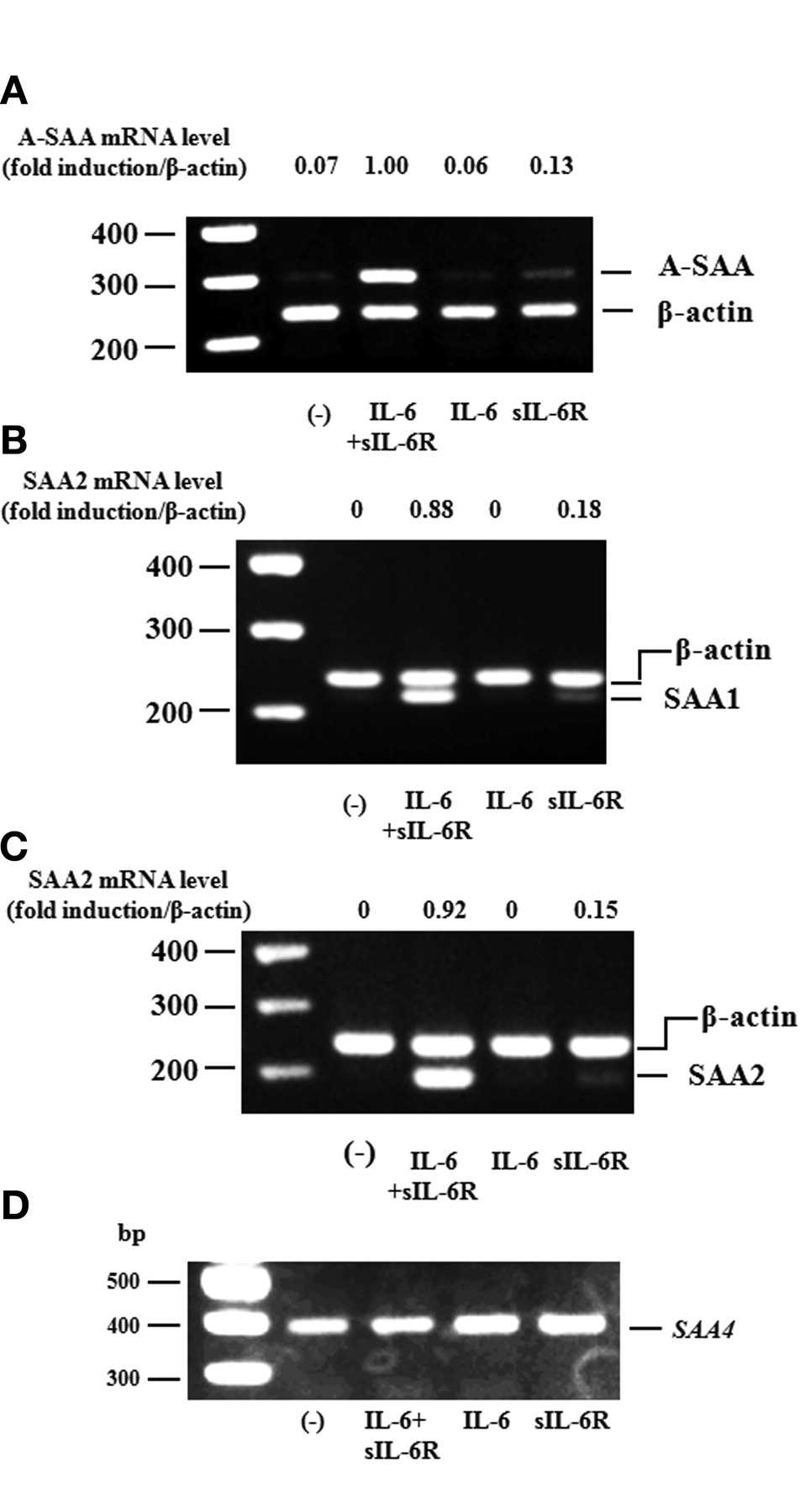
A-SAA mRNA expression on RA-FLS by IL-6. RA-FLS were stimulated with IL-6 (100 ng/ml), sIL-6R (100 ng/ml), and IL-6 + sIL-6R for 4 h. A-SAA (SAA1+2 (A), SAA1 (B), SAA2 (C), and SAA4 (D) expression were analyzed by RT-PCR. The ratio of A-SAA, SAA1, or SAA2 band against ß-actin was calculated. The data represent 3 independent experiments using different RA-FLS.
To determine the optimal conditions for activation of the A-SAA genes, we investigated the time course of A-SAA mRNA expression after IL-6 stimulation. After IL-6/sIL-6R stimulation, A-SAA was rapidly upregulated and reached a maximum at 4 h in RA-FLS (Figure 3A).

Time course or dose response of A-SAA mRNA expression by IL-6/sIL-6R. A. RA-FLS were stimulated with IL-6/sIL-6R for 2, 4, and 8 h. After culture, A-SAA mRNA expression was analyzed by RT-PCR. B. RA-FLS were stimulated with different concentrations of IL-6 for 4 h. After culture, A-SAA mRNA expression was analyzed by RT-PCR. C. RA-FLS were stimulated with different concentrations of IL-6, IL-1ß, or TNF-α for 4 h. After culture, A-SAA mRNA expression was analyzed by RT-PCR. The ratio of A-SAA band against ß-actin was calculated. The data represent 2 independent experiments using different RA-FLS.
Next, we examined whether IL-6 stimulation increased the expression of A-SAA in a dose-dependent manner. Indeed, IL-6 (4–100 ng/ml) induced A-SAA in a dose-dependent manner (Figure 3B). Twenty ng/ml of IL-6 produced a maximal activation of A-SAA; however, 100 ng/ml of IL-1ß or TNF-α were necessary for sufficient induction of A-SAA mRNA in RA-FLS (Figure 3C).
IL-6 induces activation of JAK/STAT pathways
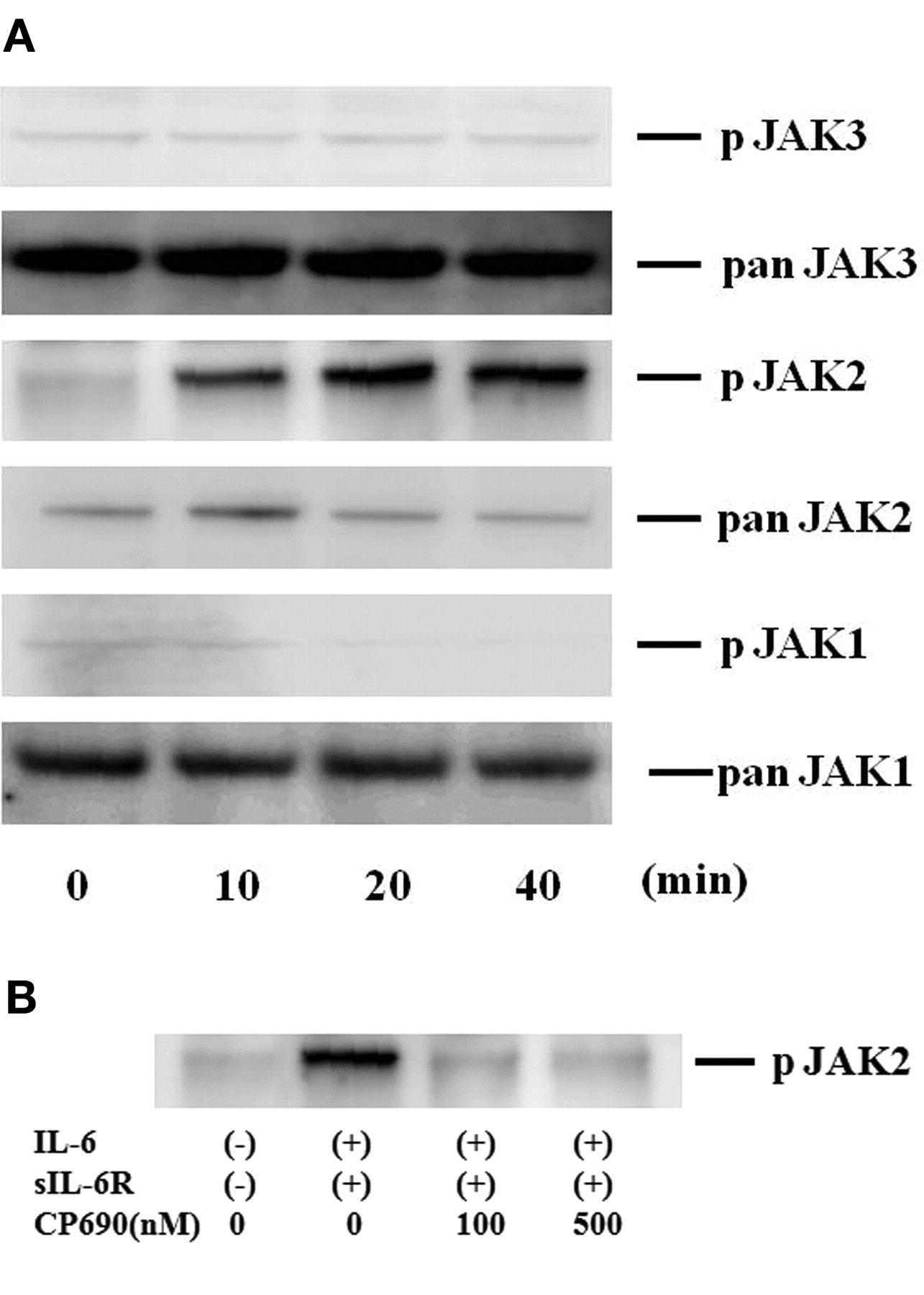
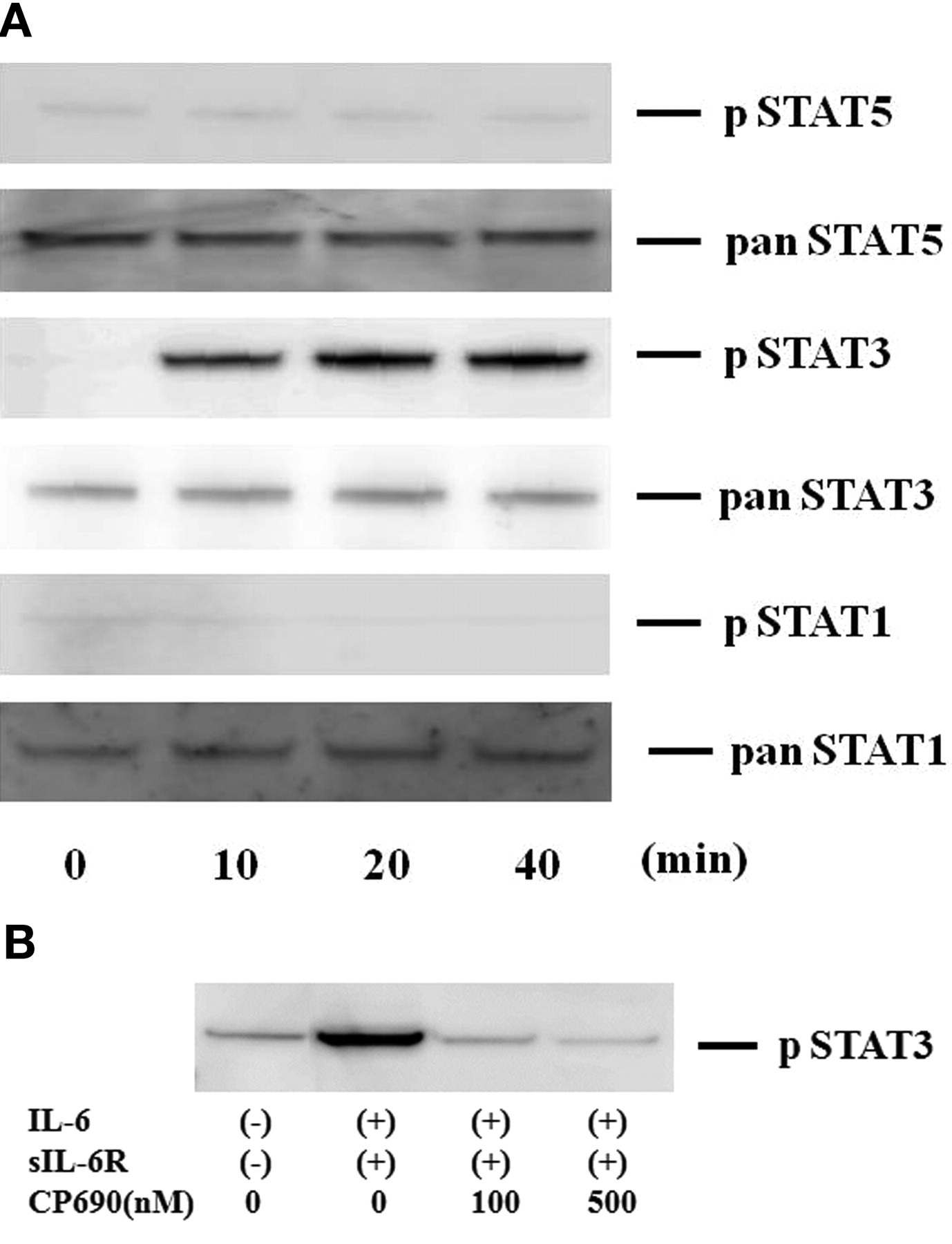
To investigate the mechanisms of IL-6-mediated signaling in the induction of A-SAA in RA-FLS, we evaluated the activation of JAK and STAT in IL-6-treated RA-FLS. Quiescent RA-FLS were stimulated with IL-6/sIL-6R for different time periods (0–40 min), and protein extracts were analyzed by immunoblotting with phospho-specific antibodies. As shown in Figure 4A, JAK2 was strongly phosphorylated after 20 min stimulation with IL-6/sIL-6R in RA-FLS. By contrast, JAK1 or JAK3 phosphorylation was not detected in IL-6/sIL-6R-stimulated RA-FLS under the same conditions. CP690,550 almost completely inhibited the IL-6/sIL-6R-induced phosphorylation of JAK2 (Figure 4B). We also examined whether STAT are phosphorylated in response to IL-6/sIL-6R stimulation in RA-FLS. As shown in Figure 5A, an increase in phospho-STAT3 protein was observed in RA-FLS after 20 min of IL-6/sIL-6R stimulation, whereas phosphorylation of either STAT1 or STAT5 was not detected in the IL-6/sIL-6R-stimulated RA-FLS under experimental conditions as described25. CP690,550 abrogated this IL-6/sIL-6R-induced phosphorylation of STAT3 in RA-FLS (Figure 5B).

CP690,550 suppressed IL6-induced JAK2 activation in RA-FLS. A. Quiescent RA-FLS were stimulated with IL-6 plus sIL-6R for indicated times. Phosphorylation of JAK1, JAK2, JAK3 was determined by Western blotting using phospho-specific or pan antibodies against JAK1, JAK2, and JAK3. B. Quiescent RA-FLS were pretreated with various concentrations of CP690,550 for 2 h, then stimulated with IL-6 plus sIL-6R for 20 min. Cellular lysates were subjected to Western blotting using phospho-specific antibodies against phospho-JAK2. Three experiments were performed using different RA-FLS: a representative result is shown.

CP690,550 suppressed IL-6-induced STAT3 activation in RA-FLS. A. Quiescent RA-FLS were stimulated with IL-6 plus sIL-6R for indicated times. Phosphorylation of STAT1, STAT3, STAT5 was determined by Western blotting using phospho-specific or pan antibodies against STAT1, STAT3, and STAT5. B. Quiescent RA-FLS were pretreated with CP690,550 for 2 h, then stimulated with IL-6 plus sIL-6R for 20 min. Cellular lysates were subjected to Western blotting using phospho-specific antibodies against STAT3. Three experiments were performed using different RA-FLS: a representative result is shown.
CP690,550 inhibits IL-6-induced A-SAA mRNA expression
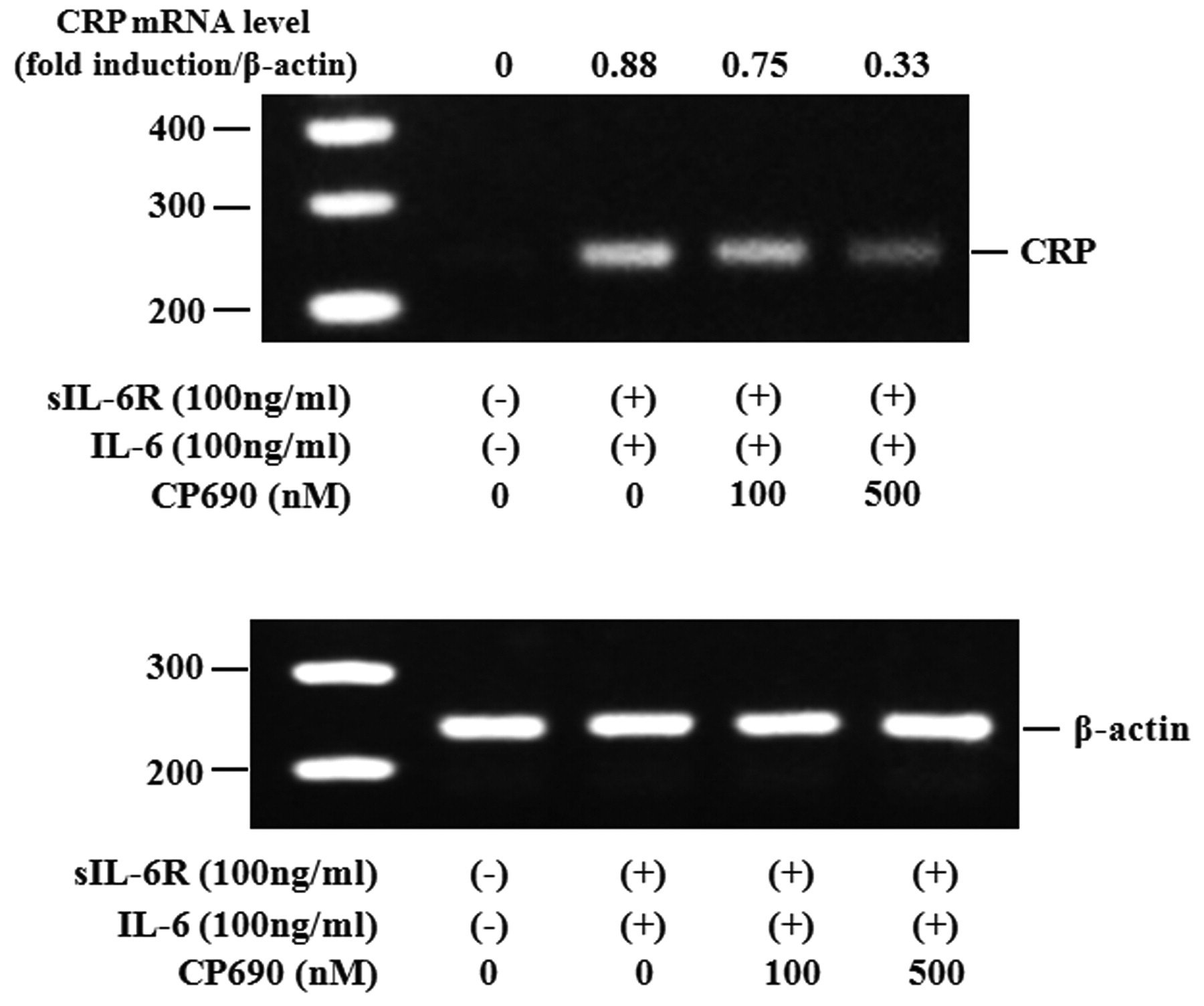
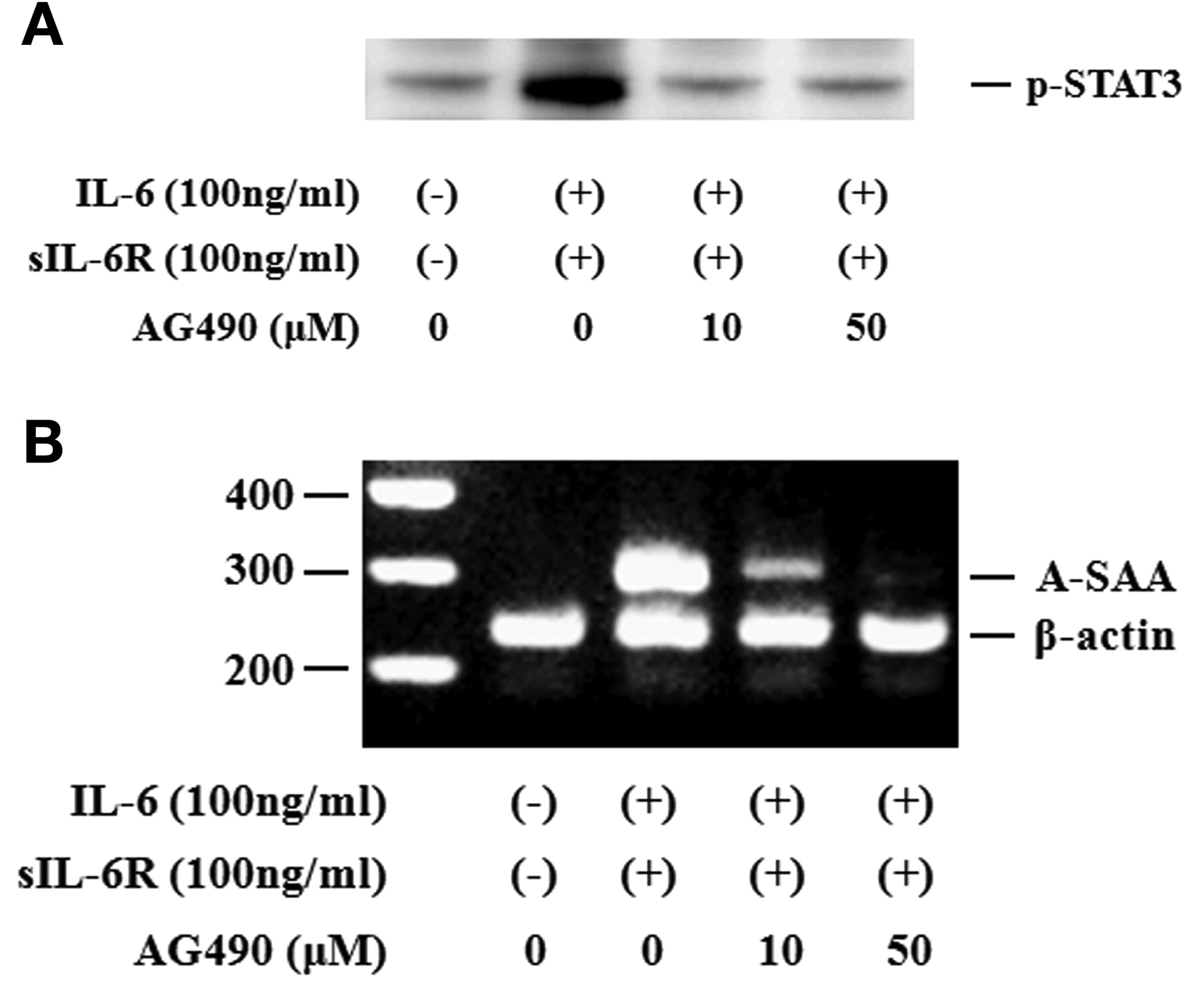
Activated STAT3 signaling has been reported to be associated with the induction of SAA. Therefore, we evaluated whether neutralizing IL-6-mediated signaling using CP690,550 affected the induction of A-SAA mRNA in RA-FLS. IL-6/sIL-6R treatment markedly elevated the expression of A-SAA mRNA in RA-FLS. Treatment with CP690,550 almost completely inhibited A-SAA mRNA expression in IL-6/sIL-6R-stimulated RA-FLS (Figure 6A, 6B). The anti-sIL-6R antibody tocilizumab also inhibited IL-6-mediated A-SAA mRNA expression in RA-FLS (Figure 6A). CP690,550 did not inhibit IL-6-induced CRP mRNA expression completely (Figure 7). Next, RA-FLS were pretreated with the JAK2 inhibitor AG490 before IL-6 stimulation. AG490 prevented IL-6-mediated STAT3 phosphorylation (Figure 8A), as well as expression of A-SAA mRNA (Figure 8B). These findings suggest that blocking the IL-6-mediated JAK2/STAT3 pathway would lead to inhibition of A-SAA mRNA expression in RA-FLS. In view of the results obtained in RA-FLS, we examined whether CP690,550 inhibited IL-6-mediated A-SAA mRNA expression in human hepatocytes. CP690,550 prevented IL-6-induced A-SAA mRNA expression (Figure 9A). However, CP690,550 partially inhibited IL-1ß-induced A-SAA mRNA expression in hepatocytes (Figure 9B).

CP690,550 inhibited IL-6-induced A-SAA mRNA expression in RA-FLS. A. RA-FLS were stimulated with IL-6 plus sIL-6R in the presence of indicated concentrations of CP690,550. A-SAA mRNA expression was analyzed by RT-PCR. Three experiments were performed using different RA-FLS: a representative result is shown. B. The ratio of A-SAA band against ß-actin was calculated and the mean ± SD of 3 independent experiments using different RA-FLS is shown. *p < 0.01, **p < 0.001 compared to IL-6/sIL-6R-treated RA-FLS.

Effects of CP690,550 on IL-6-induced CRP mRNA expression in RA-FLS. RA-FLS were stimulated with IL-6 plus sIL-6R in the presence of indicated concentrations of CP690,550. CRP mRNA expression was analyzed by RT-PCR. ß-actin mRNA expression was used as control. Two experiments were performed using different RA-FLS: a representative result is shown.
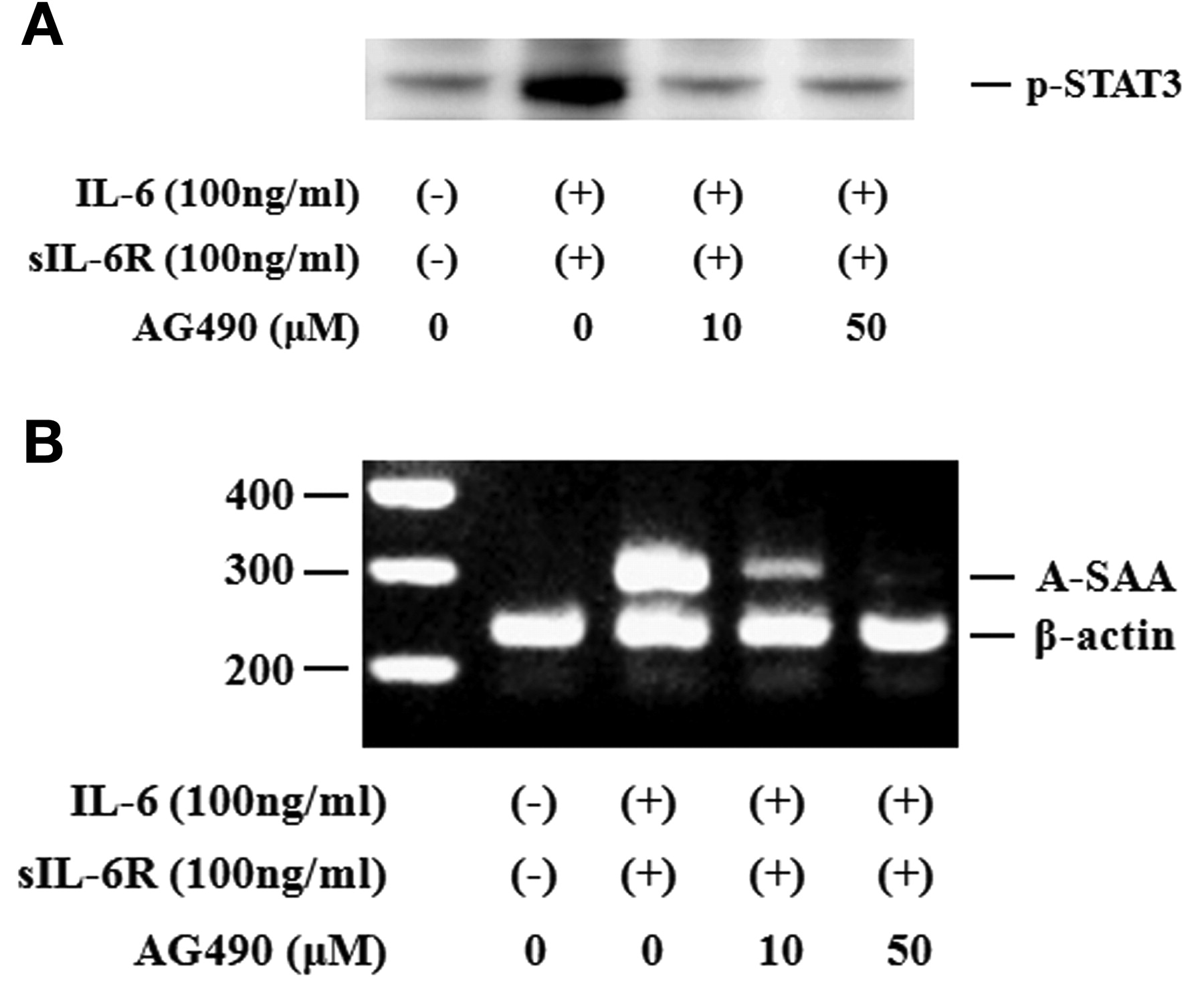
AG490 suppressed IL6-induced STAT3 activation and A-SAA mRNA expression in RA-FLS. A. Quiescent RA-FLS were stimulated with IL-6 plus sIL-6R in the presence of indicated concentrations of AG490. Phosphorylation of STAT3 was determined by Western blotting using phospho-specific antibodies against STAT3. B. RA-FLS were stimulated with IL-6 plus sIL-6R in the presence of indicated concentrations of AG490. A-SAA mRNA expression was analyzed by RT-PCR. Two experiments were performed using different RA-FLS: a representative result is shown.

Effects of CP690,550 on cytokine-induced A-SAA mRNA expression in human hepatocytes. Hepatocytes were stimulated with IL-6 plus sIL-6R (A) or IL-1ß (B) in the presence of indicated concentrations of CP690,550. A-SAA mRNA expression was analyzed by RT-PCR. Two experiments were performed using different RA-FLS: a representative result is shown.
DISCUSSION
SAA has traditionally been viewed as an acute-phase reactant and as a potential biomarker of inflammation, similar to CRP26. Recent evidence has indicated that SAA is a functionally relevant proinflammatory molecule27. SAA, which is expressed in rheumatoid synovial tissues, induces the production of cytokines or chemokines in rheumatoid synoviocytes8,28, suggesting a pathogenic role of SAA in the proinflammatory cascades of RA. One of the major factors thought to contribute to the induction of SAA in RA patients is IL-629. Activation of rheumatoid synoviocytes by IL-6 may regulate the local induction of SAA, which may contribute to the inflammatory synovial environment of RA. Indeed, monoclonal antibodies that block IL-6R-mediated signaling have been proven to be clinically beneficial by normalizing the elevated circulating levels of SAA in patients with RA14. IL-6 signaling is initiated by IL-6 binding with the IL-6R complex containing the signal transducer glycoprotein 130 (gp130)30. This results in the activation of JAK, by phosphorylating gp130, which results in the recruitment of STAT family members, mainly STAT331. JAK-mediated phosphorylation of STAT3 is crucial for its nuclear translocation and for activating transcription of downstream target genes such as SAA32.
Although diverse roles of each STAT family member have been suggested in RA, the JAK/STAT signal transduction pathways are activated in rheumatoid synovium by hematopoietic cytokine families such as IL-733. We investigated the IL-6 signaling pathway using RA-FLS. Our data indicated that IL-6/IL-6R signaling induced the expression of A-SAA mRNA in RA-FLS, and that JAK2 and STAT3 activation are involved in this IL-6-mediated induction of A-SAA.
Another aim of our study was to elucidate the role of JAK inhibition in the IL-6-dependent pathway that leads to induction of A-SAA in RA-FLS. We demonstrated that IL-6-stimulated activation of JAK2 and STAT3 was abrogated by CP690,555 at physiological concentrations in RA-FLS. Further, CP690,550 inhibited the IL-6-induced expression of A-SAA (SAA1 + SAA2) mRNA in RA-FLS. Our data suggest that the JAK2/STAT3 pathway is mainly activated in the IL-6 trans-signaling pathway in RA-FLS and that this pathway was blocked by CP690,550. CP690,550 was originally believed to be a JAK3 inhibitor34. However, it has now become clear that in addition to its effect on JAK3 this compound inhibits JAK1 and JAK2 with similar IC50 values35,36. Therefore, it is possible that CP690,550 inhibits the IL-6-induced JAK2 and subsequent STAT3 activation. Selective JAK3 inhibition has theoretical advantages in immunosuppression, since JAK3 expression is restricted to immune cells. Dendritic cells in rheumatoid synovium were found to express JAK3; therefore, it is presumed that CP690,550 potently inhibits cytokine-receptor signaling pathways of immune cells in rheumatoid synovium37. Despite the excellent selectivity to JAK3, CP690,550 was recently reported to have modest selectivity against JAK1 and JAK221. Indeed, our data indicate that CP690,550 interferes with IL-6-mediated SAA induction in nonimmune RA-FLS, which could be explained by the effects of the JAK2/STAT3 pathway. A specific JAK2 inhibitor, AG490, also prevented IL-6-mediated SAA induction, suggesting that the JAK2/STAT3 pathway contributes to IL-6 signaling in RA-FLS.
Hagihara, et al demonstrated that STAT3 plays an essential role in IL-1 + IL-6-induced expression of the A-SAA genes18. While human SAA genes do not exhibit the typical STAT3 response element (RE) in their promoters, they demonstrated that STAT3 and nuclear factor-κB (NF-κB) p65 form a complex that interacts with the 3′ site of the NF-κB RE in the promoters of the SAA genes. They concluded that STAT3 forms a complex with NF-κB and contributes to transcriptional activation of A-SAA genes18. Pietrangelo, et al showed that IL-6-mediated induction of SAA1 and SAA2 was eliminated in conditional STAT3 knockout mice38. According to these reports, STAT3 might be the main mediator in the activation of A-SAA. JAK phosphorylate STAT3, which is crucial for the nuclear translocation of STAT3 and for activation of transcription of its downstream targets, such as SAA. We observed that the IL-6-stimulated activation of STAT3 was repressed by CP690,550, probably by blockade of the upstream signaling molecule, JAK2. Therefore, inhibition of STAT3 could be a mechanism by which CP690,550 exerts its antiinflammatory effects. From our data, IL-6 was a more potent inducer for SAA compared to IL-1ß or TNF-α in RA-FLS. Also, previous reports indicated that either IL-6 and IL-1ß or IL-6 and TNF-α, but not IL-1ß and TNF-α, induced the synergistic induction of SAA1 and SAA2 gene expression in human hepatocytes15. These findings suggest that IL-6 plays a critical role in the synergistic induction of human SAA gene by stimulation with inflammatory cytokines. TNF-α and IL-1ß induce A-SAA, which mainly is dependent on the NF-κB pathway39. The pathogenic role of TNF-α in AA amyloidosis was suggested from a report of the efficacy of anti-TNF-α therapy against AA amyloidosis secondary to inflammatory arthritis40. While the influence of TNF-α on the JAK/STAT pathway is currently unknown, TNF-α blocker might not only reduce the synthesis of SAA but also slow amyloid deposition by affecting the expression of receptors for advanced glycation endproducts that are involved in amyloid fibril deposition41.
SAA is the precursor of amyloid A protein in AA amyloidosis3. Normalization of serum SAA levels can lead to amyloid regression in patients with AA amyloidosis42,43. Indeed, anti-IL-6R antibody treatment improves the clinical symptoms of patients with RA who have AA amyloidosis44. One of the major factors contributing to the induction of SAA in patients with RA is IL-615. Activation of RA-FLS by IL-6 trans-signaling may regulate the local induction of SAA that may contribute to the synovial inflammatory environment of RA. Locally produced SAA contributes to rheumatoid synovitis by stimulating MMP synthesis, as described45. Therefore, blocking IL-6 signaling using CP690,550 may silence the rheumatoid synovitis by inhibiting induction of SAA.
The complex interplay of different transcriptional factors and signaling molecules on A-SAA induction during IL-6 stimulation remains to be completely elucidated; however, our data suggest that STAT3 plays an important role in the IL-6/A-SAA axis in RA-FLS. Inhibition of this axis using CP690,555 could represent a new therapeutic strategy for rheumatoid inflammation and AA amyloidosis in patients with RA.
Footnotes
-
Supported by a Grant-in-Aid for Scientific Research of the Japan Society for the Promotion of Science.
- Accepted for publication May 30, 2011.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}