

To the Editor:
Catastrophic antiphospholipid antibody syndrome (CAPS) was initially described in 1992 by Asherson1. The syndrome occurs in a very small proportion of patients with APS and carries significant morbidity and mortality. It is defined as acute multiorgan failure resulting from multiple, rapidly developing vascular occlusions. The main organs involved are the renal, pulmonary, and nervous systems. Other organs, including skin, heart, adrenal glands, and liver can also be affected. Rarely, CAPS is associated with bone marrow involvement and necrosis due to microvascular thrombosis2. This case details that rare relationship and the need for early identification and treatment of CAPS.
A 40-year-old Hispanic woman presented to the emergency department with 5 hours of severe lumbosacral pain. The pain developed suddenly and was aggravated by movement. She related that the pain radiated to the left lower extremity. There was no history of trauma and she found that nothing alleviated the pain. Her history was significant for chronic thrombocytopenia, menorrhagia (for which she was taking oral contraceptives), and 2 second-trimester spontaneous abortions. Examination revealed paraspinal tenderness in the lumbosacral region. Plain radiographs of lumbosacral spine demonstrated only a loss of normal lordosis.
The white blood cell count was 6200/μl, with 85% neutrophils, 11% lymphocytes, 14% monocytes, and 2% eosinophils. Her hemoglobin was 6.7 g/dl and her mean corpuscular volume was 74/μm3. Her platelet count at the time of admission was 48,000/μl. The peripheral smear revealed no evidence of schistocytes. A computed tomography scan was unremarkable and followup magnetic resonance imaging (MRI) was recommended. The MRI showed decreased enhancement at L2, L3, and L5 (Figure 1). This was followed by a bone scan showing decreased uptake in the same areas (Figure 2). This combination of findings is consistent with bone marrow necrosis (BMN).
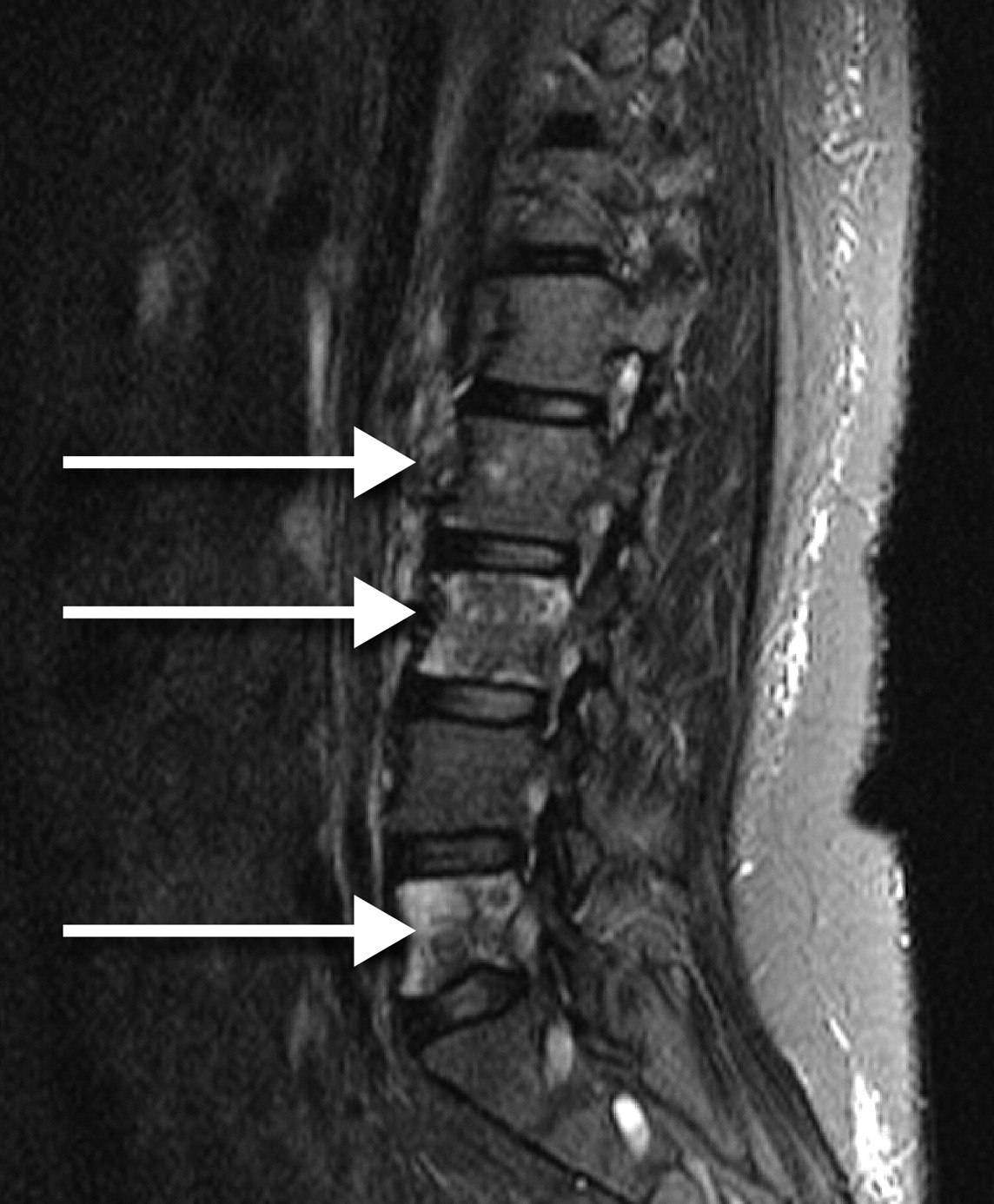
Magnetic resonance imaging of the lumbar spine showed decreased enhancement at L2, L3, and L5 (arrows).

Bone scan showed decreased uptake (arrows).
On the patient’s fourth hospital day, her platelet count dropped to 12,000/μl and prednisone was initiated at a dose of 40 mg twice daily. The platelet count subsequently increased to 30,000/μl within a day. She later became acutely hypoxemic and hypotensive, and a chest radiograph demonstrated bilateral pulmonary edema. A repeat platelet count was 6000/μl and her activated partial thromboplastin time rose to 111.5 s, with a protime of 13.7 s and an international normalized ratio of 1.2. Her amylase and lipase increased to 2530/μl and 428/μl, respectively.
Pulmonary specialists were consulted because of her new chest radiograph findings and deteriorating clinical condition. Cardiogenic pulmonary edema was unlikely given the lack of examination findings consistent with heart failure syndrome and a normal transthoracic echocardiogram. The primary concerns were the acute respiratory distress syndrome and intraalveolar hemorrhage, the latter primarily because of the patient’s severe thrombocytopenia. Bronchoscopy was performed and revealed no evidence of intraalveolar hemorrhage, and she was subsequently intubated and broad-spectrum antibiotic coverage was initiated.
Concurrent with her pulmonary deterioration, she developed acute renal failure as her creatinine rose from 0.9 mg/dl at admission to 4.1 mg/dl. A 24-hour urine collection revealed 2.5 g proteinuria with innumerable red blood cells. Given these findings, c-antineutrophil cytoplasmic autoantibodies and p-antineutrophil cytoplasmic antibodies and antiglomerular basement membrane antibodies were ordered. These serologies were negative but an antinuclear antibody was positive and a rheumatologist was subsequently consulted. The rheumatologist began the patient on 1 g methylprednisolone daily for 3 days and ordered additional serologic testing. A positive antiphospholipid antibody (aPL) profile was confirmed with a positive anticardiolipin antibody (aCL) IgG at 37.5, lupus anticoagulant (LAC), Russell viper venom (RVV) test, and apolipoprotein H (APOH).
A presumptive diagnosis of CAPS, or Asherson’s syndrome, was made, and a confirmatory renal biopsy revealed thrombotic microangiopathy. High-dose corticosteroids and pheresis platelet transfusion had no effect on the platelet count. The patient received intravenous immunoglobulin (IVIG) for 5 days, increasing the platelet count to 140,000/μl. Sputum cultures grew Klebsiella pneumoniae, and the antibiotic coverage was narrowed to cefotaxime monotherapy, based upon sensitivity reporting.
The clinical condition improved and she was ultimately extubated. Her mental status gradually improved to baseline without any residual neurological deficit. Renal function, amylase, and lipase returned to baseline within 2 weeks. The back pain resolved and she was discharged on a tapering dose of prednisone. Unfortunately, the patient was lost to followup and no further laboratory studies were obtained to evaluate her serum antibodies over time.
This patient’s clinical experience is consistent with BMN resulting from CAPS-induced microvascular thrombosis. She had multiorgan involvement secondary to this small-vessel ischemia and there were several predisposing factors for CAPS. The use of oral contraceptives while having undiagnosed APS and respiratory infection with gram-negative bacteria (Klebsiella) were multiple triggers for this patient3. About 0.9% of patients with APS develop CAPS4. Precipitating factors are found in 55% of cases and are most frequently attributed to infectious causes. The mortality rate associated with CAPS approaches 50%4,5. It is likely unrecognized in many intensive care units and should be considered in patients with multiorgan failure of unknown origin.
Classification criteria have been developed for CAPS (Table 1)6. A definitive diagnosis of CAPS requires all 4 criteria to be present, and this patient did meet each criterion. This patient was fortunate to recover with the best-known therapy for CAPS, which includes treatment of any identified triggers, systemic anticoagulation, high-dose corticosteroids, and IVIG. Clinical improvement occurred rapidly after the addition of high-dose corticosteroids and IVIG.
Criteria for catastrophic antiphospholipid antibody syndrome.
BMN is a relatively uncommon condition and is most frequently encountered in patients with malignancy (metastatic disease and leukemia), direct injury to the bone marrow (e.g., radiation therapy), infection (human immunodeficiency virus and septic shock), and sickle-cell disease. Studies have established aPL as a cause of marrow necrosis7,8. This patient had elevated titers of aPL (aCL, LAC, RVV, and APOH) and it is believed that the higher levels of antibodies are necessary to cause BMN.
CAPS is a life-threatening complication of APS that requires prompt identification and treatment because of its high mortality rate. BMN is a rare manifestation of CAPS, caused by microvascular thrombosis associated with CAPS. This uncommon finding must be recognized as a complication of CAPS to allow identification of the underlying syndrome.








{kind=link}
{kind=link}