
To the Editor:
Extramusculocutaneous manifestations in juvenile dermatomyositis (JDM) may lead to life-threatening consequences. Interstitial lung disease (ILD) has been reported as one such serious complication in JDM1,2,3,4; how ever, cardiac involvement in JDM is a rare complication and is seldom reported5,6. Recently, anti-CADM-140 autoantibody was discovered in amyopathic dermatomyositis and was associated with rapidly progressive ILD7,8. We describe a fatal case of JDM complicated by ILD and cardiac involvement in which serum preserved at admission was shown to contain anti-CADM-140 antibody.
A 9-year-old boy was admitted to our hospital with a 4-month history of low-grade fever and erythematous rashes on his face, hands, elbows, and knees. He was developmentally delayed from an unknown cause. He could not describe muscle weakness or tenderness but showed claudication indicating lower-limb muscle weakness. He had Gottron’s papules but no heliotrope rash. Cardiac sounds revealed a gallop rhythm and he had fine crackles over both lung fields. His erythrocyte sedimentation rate was 38 mm/h, white cell count 1800/μl, hemoglobin 9.3 g/dl, platelets 115,000/μl, aspartate aminotransferase 207 U/l (normal 11–39), alanine aminotransferase 101 U/l (normal 5–40), lactate dehydrogenase 564 U/l (normal 119–229), aldolase 16.7 U/l (normal 2.1–6.1), creatine kinase (CK) 104 U/l (normal 45–160), C-reactive protein (CRP) 0.24 mg/dl (normal 0.0–0.3), antinuclear antibody 80 (normal < 40), anti-Jo-1 antibody negative, brain natriuretic peptide 55.2 pg/ml (normal 0.0–19.5), and Krebs von den Lungen-6 (KL-6) 1275 U/ml (normal 0–500). His electrocardiogram revealed sinus tachycardia at 150 beats/min and T wave flattening. Echocardiography demonstrated a low ejection fraction of 44%. Chest high-resolution computed tomography (HRCT) revealed bilateral pleural effusions. Whole-body scintigraphy with 67Ga-citrate showed increased uptake in the lower lobes of the lungs. Electromyography showed low amplitude and short durations compatible with myopathy. Magnetic resonance imaging showed increased signals at bilateral adductor and pectineal muscles on T2-weighted images. He was diagnosed with JDM and initially treated with oral methylpredonisolone (mPSL). However, his CK increased to 315 U/l two weeks later and two courses of mPSL pulse therapy were added. After each mPSL pulse therapy ejection fraction was temporarily normalized. One month later the pleural effusions had resolved but new infiltrations developed at the base of both lower lobes on HRCT. We started monthly intravenous cyclophosphamide pulse therapy. Two weeks later, however, fever developed and CRP increased to 4.60 mg/dl. Bacterial cultures from blood, sputum, and aspirated pleural effusion were negative. Serum ß-D-glucan and cytomegalovirus antigen were also negative. Pneumocystis jiroveci was not detected in aspirated pleural effusion. Repeat HRCT demonstrated progressive bilateral infiltration in the lower lobes, then intravenous cyclosporin A was administered. KL-6 was elevated to 5138 U/ml. His respiratory condition rapidly deteriorated and pneumomediastinum, subcutaneous emphysema, and left pneumothorax developed (Figure 1). He was intubated and mechanically ventilated and underwent plasmapheresis. Despite intensive treatment, he died 1 week after intubation, which was 3 months after the initial admission to hospital.
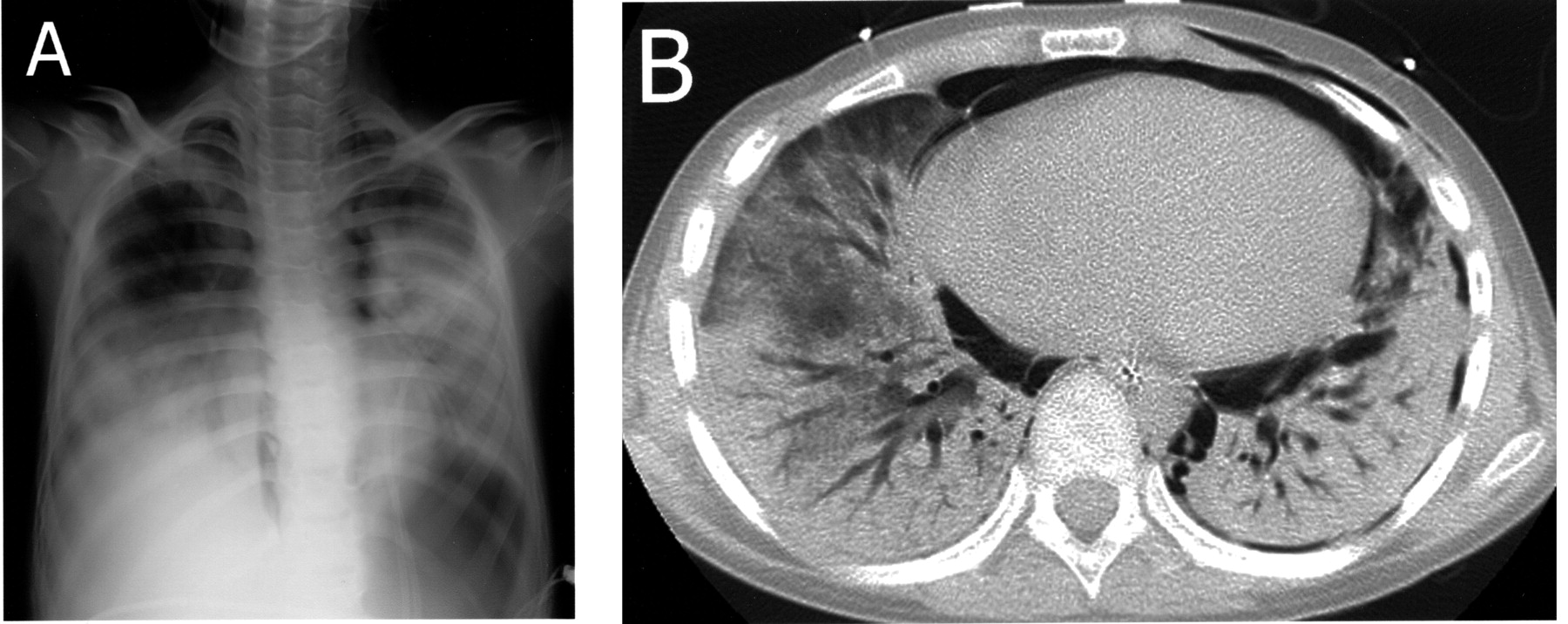
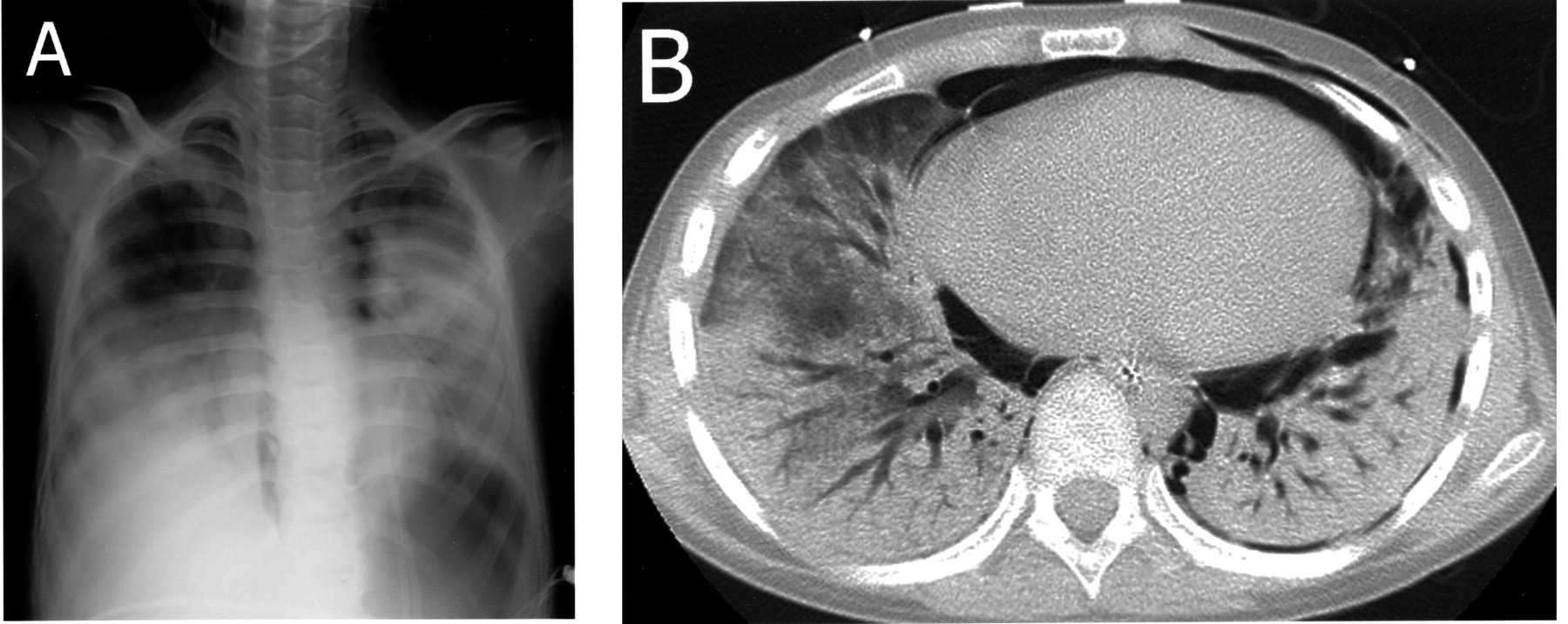
A. Chest radiography showing diffuse bilateral consolidation of the lungs with pneumomediastinum and subcutaneous emphysema of thoracic walls. B. Chest HRCT shows extensive bilateral lobar consolidation, pneumomediastinum, subcutaneous emphysema of the thoracic walls, and left pneumothorax.
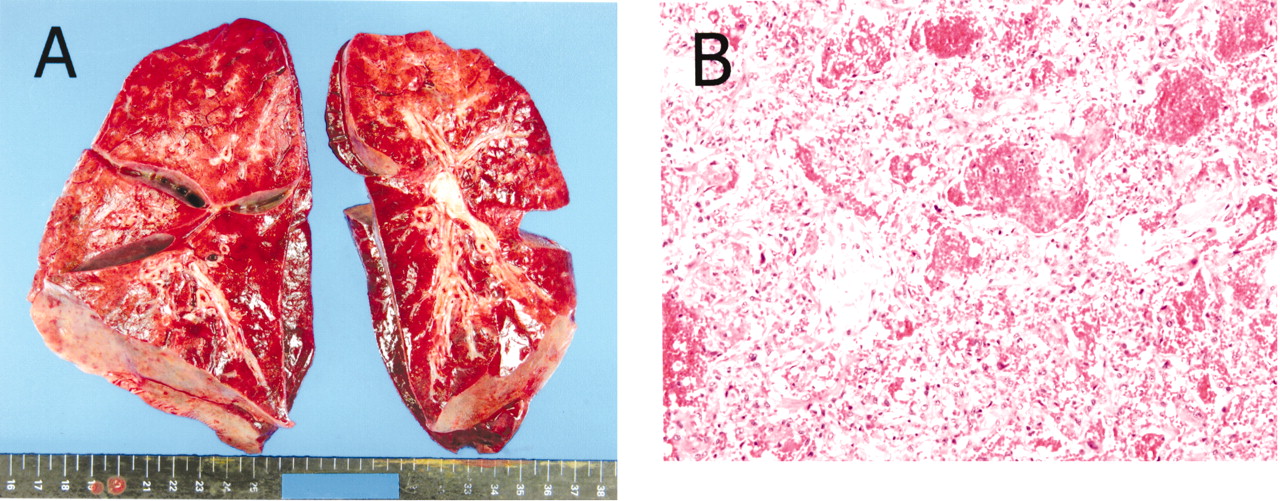
With his guardian’s consent, an autopsy was performed. The lungs were entirely firm and showed remarkable congestion, and the microscopic examination revealed intense alveolar hemorrhage, formation of hyaline membrane, and infiltration of inflammatory cells suggesting diffuse alveolar damage (Figure 2). Neither interstitial changes nor infection of the lungs were present. The heart demonstrated hypertrophy and histologic examination detected no specific changes of myocarditis, pericarditis, endocarditis, or fibrosis except for partial degeneration of the papillary muscles of the left ventricle. Serum anti-CADM-140 antibody was measured by ELISA in his serum preserved at admission (courtesy of Dr. Masataka Kuwana, Keio University of Japan)7,8. Anti-CADM-140 antibody was elevated (25.256 units, normal below 8).
{kind=link}
{kind=link}
Autopsy findings of the lungs. A. Severe congestion of the lungs. B. The lung histology shows alveolar hemorrhage, formation of hyaline membrane, and infiltration of inflammatory cells compatible with diffuse alveolar damage (H&E stain, original magnification ×200).
ILD is a rare but serious complication of JDM1,2,3,4. In adult cases of polymyositis (PM) and dermatomyositis (DM), histologic findings of ILD have been shown to be correlated with clinical outcomes9,10. In particular, cases of PM/DM with diffuse alveolar damage have shown the worst prognosis among those with major histopathology of ILD, as observed in our case9,10. Our patient had impaired cardiac function during his admission. At autopsy, partial degeneration of the papillary muscles of the left ventricle was the only finding. It is unclear whether this histologic change is sufficient to explain his impaired cardiac function. In previous reports, the cardiac features seen most often in cases of PM/DM/JDM were myocarditis, myocardial fibrosis, and vascular abnormalities on histologic examination, features not seen in our case5,6.
Anti-CADM-140 antibody was recently discovered in clinically amyopathic patients with DM and its presence was associated with rapidly progressive ILD7,8. The presence of anti-CADM-140 antibody in our patient may suggest that this antibody is a predictor of rapidly progressive ILD and poor prognosis even in JDM.
We describe a case of fatal JDM with rapidly progressive ILD and cardiac involvement. Positive anti-CADM-140 antibody may predict a poor prognosis for JDM with ILD.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.