Article Text

Abstract
The exact patho-aetiology of systemic lupus erythematosus (SLE) remains elusive. An extremely complicated and multifactorial interaction among various genetic and environmental factors is probably involved. Multiple genes contribute to disease susceptibility. The interaction of sex, hormonal milieu, and the hypothalamo–pituitary–adrenal axis modifies this susceptibility and the clinical expression of the disease. Defective immune regulatory mechanisms, such as the clearance of apoptotic cells and immune complexes, are important contributors to the development of SLE. The loss of immune tolerance, increased antigenic load, excess T cell help, defective B cell suppression, and the shifting of T helper 1 (Th1) to Th2 immune responses leads to B cell hyperactivity and the production of pathogenic autoantibodies. Finally, certain environmental factors are probably required to trigger the disease.
- aetiology
- pathogenesis
- genetic
- interaction
- autoimmune
- autoantibody
- APC, antigen presenting cell
- CD40L, CD40 ligand
- CRH, corticotrophin releasing hormone
- DHEA, dehydroepiandrosterone
- dsDNA, double stranded DNA
- FasL, Fas ligand
- GnRH, gonadotrophin releasing hormone
- HLA, human leucocyte antigen
- HPA, hypothalamo–pituitary–adrenal
- HRT, hormonal replacement therapy
- IL, interleukin
- LH, luteinising hormone
- MBP, mannose binding protein
- MHC, major histocompatibility complex
- NK, natural killer
- nRNP, nuclear ribonuclear protein
- OC, oral contraceptive
- PBMC, peripheral blood mononuclear cell
- SLE, systemic lupus erythematosus
- sm, Smith antigen
- snRPN, small nuclear ribonuclear protein
- Th1, T helper type 1
Statistics from Altmetric.com
- APC, antigen presenting cell
- CD40L, CD40 ligand
- CRH, corticotrophin releasing hormone
- DHEA, dehydroepiandrosterone
- dsDNA, double stranded DNA
- FasL, Fas ligand
- GnRH, gonadotrophin releasing hormone
- HLA, human leucocyte antigen
- HPA, hypothalamo–pituitary–adrenal
- HRT, hormonal replacement therapy
- IL, interleukin
- LH, luteinising hormone
- MBP, mannose binding protein
- MHC, major histocompatibility complex
- NK, natural killer
- nRNP, nuclear ribonuclear protein
- OC, oral contraceptive
- PBMC, peripheral blood mononuclear cell
- SLE, systemic lupus erythematosus
- sm, Smith antigen
- snRPN, small nuclear ribonuclear protein
- Th1, T helper type 1
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease characterised by the production of antibodies to components of the cell nucleus in association with a diverse array of clinical manifestations. The primary pathological findings in patients with SLE are those of inflammation, vasculitis, immune complex deposition, and vasculopathy. The exact aetiology of SLE is unknown. SLE shows a strong familial aggregation, with a much higher frequency among first degree relatives of patients. Moreover, in extended families, SLE may coexist with other organ specific autoimmune diseases such as haemolytic anaemia, immune thrombocytopenic purpura, and thyroiditis. The concordance of the disease in identical twins is approximately 25–50% and that in dizygotic twins is around 5%.1 This suggests that genetic factors play an important role in the predisposition of the disease. However, most cases of SLE are sporadic without identifiable genetic predisposing factors, suggesting that multiple environmental or yet unknown factors may also be responsible.
GENETIC SUSCEPTIBILITY
The concordance of SLE in identical twins, the increase in frequency of SLE among first degree relatives, and the increased risk of developing the disease in siblings of SLE patients reflects a polygenic inheritance of the disease. Many different genes contribute to disease susceptibility. In a small proportion of patients (< 5%), a single gene may be responsible. For instance, patients with homozygous deficiencies of the early components of complement are at risk of developing SLE or a lupus-like disease.2 For most of the remaining patients, multiple genes are required. It is estimated that at least four susceptibility genes are needed for the development of the disease.3
Of the genetic elements, the genes of the major histocompatibility complex (MHC) have been most extensively studied for their contribution to human SLE. Population studies reveal that the susceptibility to SLE involves human leucocyte antigen (HLA) class II gene polymorphisms. An association of HLA DR2 and DR3 with SLE is a common finding in patients of different ethnicities, with a relative risk for the development of disease of approximately two to five.1 The HLA class II genes have also been associated with the presence of certain autoantibodies such as anti-Sm (small nuclear ribonuclear protein), anti-Ro, anti-La, anti-nRNP (nuclear ribonuclear protein), and anti-DNA antibodies.3
“It is estimated that at least four susceptibility genes are needed for the development of the disease”
Among other MHC gene systems, inherited complement deficiencies also influence disease susceptibility. The HLA class III genes, particularly those encoding complement components C2 and C4, confer risk for SLE in certain ethnic groups. Patients with homozygous C4A null alleles, irrespective of the ethnic background, are at high risk of developing SLE. Moreover, SLE is associated with inherited deficiencies of C1q, C1r/s, and C2.4 A decrease in complement activity could promote disease susceptibility by impairing the neutralisation and clearance of self and foreign antigens. When the antigen burden overwhelms the clearance capacity of the immune system, autoimmunity may ensue.
In addition, many polymorphic non-MHC genes have been reported to be associated with SLE. These include genes that encode mannose binding protein (MBP), tumour necrosis factor α, the T cell receptor, interleukin 6 (IL-6), CR1, immunoglobulin Gm and Km allotypes, FcγRIIA and FcγRIIIA (both IgG Fc receptors), and heat shock protein 70.3,5 However, in most cases, consistent results could not be obtained in subsequent studies in different ethnic groups. Some of these polymorphic genes may confer risk to certain subsets of patients with SLE. For instance, the FcγRIIA polymorphism has been associated with nephritis in African Americans and Koreans,6,7 and the FcγRIIIA polymorphism with SLE in Hispanics and white populations.8,9 In addition, mutations of codon 54 of the MBL gene carry a minor risk for SLE susceptibility in southern Chinese.10
During the past few years, linkage analyses using SLE multiplex families have provided many chromosomal regions for further exploration of susceptibility genes.11–14 Six regions exhibiting significant linkage to SLE are promising. Studies are under way to fine map these linked regions and to identify the genes in the susceptibility regions. The discovery of multiple chromosome regions conferring risk for SLE development supports the notion that SLE is a polygenic disease. Table 1 summarises the genes that are involved in human SLE.
Genes involved in human systemic lupus erythematosus
SEX, HORMONES, AND THE HYPOTHALAMO–PITUITARY–ADRENAL AXIS
SLE is predominantly a female disease.15 First onset of SLE before puberty and after menopause16 is uncommon. The female predilection becomes less pronounced outside the reproductive age range. In addition, patients with Klinefelter’s syndrome, characterised by hypergonadotrophic hypogonadism, are prone to the development of SLE.17 These observations suggest a role for endogenous sex hormones in disease predisposition.
Abnormal oestrogen metabolism has been demonstrated in patients with SLE of both sexes, with an increase in 16α hydroxylation of oestrone, resulting in significantly raised 16α hydroxyestrone concentrations.18 The 16α metabolites are more potent and feminising oestrogens. Women with SLE also have low plasma androgens, including testosterone, dihydrotestosterone, dehydroepiandrosterone (DHEA), and dehydroepiandrosterone sulfate.19,20 This abnormality might be explained by increased testosterone oxidation at C-1721 or increased tissue aromatase activity.22 The concentrations of androgens correlate inversely with disease activity.20 Low concentrations of plasma testosterone and raised luteinising hormone (LH) values23,24 have been found in some men with SLE. Thus, excessive oestrogenic but inadequate androgenic hormonal activity in both men and women with SLE might be responsible for the alteration of the immune responses.
Table 2 summarises the actions of oestrogens on various cell types of the immune system. Both physiological and supraphysiological concentrations of oestrogens facilitate humoral responses, leading to increased B cell proliferation and antibody production.25–28 On the contrary, high doses of oestrogens inhibit T cell responses, such as proliferation and IL-2 production.29,30 Oestrogens also increase calcineurin mRNA levels and enhance the cell surface expression of CD40 ligand (CD40L) in cultured T cells from patients with SLE.31,32 These effects appear to be unique to patients with SLE, indicating that lupus T cells are more sensitive to oestrogens. Taken together, oestrogens may aggravate SLE by prolonging the survival of autoimmune cells, increasing T helper type 2 (Th2) cytokine production, and stimulating B cells to produce autoantibodies. The inhibition of the Th1 response and the enhancement of CD40L expression on lupus T cells may indirectly promote the Th2 response and lead to further B cell hyperactivity.
The effect of androgens on lymphocyte functions has been less well studied. Testosterone reduces immunoglobulin production from peripheral blood mononuclear cells of both normal subjects and patients with SLE.33,34 DHEA has been shown to be associated with enhancement of Th1 and inhibition of Th2 immune responses in both humans and mice.35,36
The opposite effects of oestrogens and androgens on the immune system, coupled with unbalanced oestrogenic and androgenic hormonal activity in patients with SLE, may help explain some of the immune aberrations seen in this disease.
Epidemiological studies reveal an association between the use of exogenous oestrogens and the onset of SLE. In a large cohort of nurses as part of the Nurses’ Health Study, it was shown that both the past use of oral contraceptive (OC) pills and hormonal replacement therapy (HRT) was associated with a slightly increased risk of SLE development.37,38 Moreover, a proportional increase in the risk of SLE related to the duration of HRT was seen. A more recent case–control study also reported an increased risk of the development of SLE or discoid lupus in women receiving HRT for more than two years when compared with non-users.39
There is evidence that endogenous oestrogen concentrations may influence disease activity and prognosis in human SLE. In the presteroid era, improvement of SLE was noted in individual patients who had undergone menopause or oophorectomy.40 Flares of SLE are well known to occur during periods of rapid hormonal changes. These include pregnancy, puerperium, ovulation stimulation during in vitro fertilisation, and exogenous oestrogen administration.41–44 Lupus activity tends to be reduced when patients undergo menopause.45 It has also been noted that in many women disease flares are more common during the second half of the menstrual cycle, after the midcycle surge of oestrogen.46 The administration of exogenous oestrogens in the form of OC pills and HRT may exacerbate the disease in patients with existing lupus.47–49 Furthermore, late onset SLE, defined as first onset of disease after the age of 50, was reported have a more benign disease course, with less serious organ involvement.50 Patients with SLE who have low female sex hormone values at disease onset were found to have a lower relative mortality risk when compared with age matched controls.51
Prolactin has recently been found to be an immunostimulatory hormone.52 The main origin is the anterior pituitary, but lymphocytes are also capable of producing prolactin, which serves as an autocrine or paracrine mediator.53 Chronic hyperprolactinaemia induced by syngeneic pituitary gland implantation stimulates primary humoral antibody responses in rats,54 and accelerates autoimmune phenomena in lupus prone mice.55 Recent data suggest that the stimulatory actions of oestrogens on autoreactive B cells require the presence of prolactin.56
Hyperprolactinaemia has been demonstrated in a proportion of patients with SLE of both sexes.57–64 Prolactin concentrations correlate with disease activity in some studies.58,64 Bromocriptine, a dopamine agonist that selectively inhibits prolactin secretion from the pituitary, has been shown to be useful in the treatment of non-life threatening SLE.65,66 However, the exact role of prolactin in SLE requires further work because a positive correlation between lupus activity and prolactin values cannot be demonstrated consistently.59,62–64
“There is evidence that endogenous oestrogen concentrations may influence disease activity and prognosis in human systemic lupus erythematosus”
Gonadotrophin releasing hormone (GnRH), a decapeptide produced by the hypothalamus, regulates the release of LH and follicle stimulating hormone from the anterior pituitary. Recent animal studies show that GnRH is immunostimulatory.67 In lupus prone mice, GnRH has been shown to exacerbate lupus, but the effect appears to be sexually dimorphic.68,69 However, the role of GnRH in human SLE requires further evaluation.
The hypothalamo–pituitary–adrenal (HPA) axis is the chief component of the stress system. The stress induced increase in serum concentrations of glucocorticoids is essential for the prevention of autoreactive or unrestrained amplification of the immune response, which results in self injury and autoimmunity. A defective HPA axis may confer susceptibility to autoimmune disorders. Female Lewis (LEW/N) rats, characterised by a defective hypothalamic corticotrophin releasing hormone (CRH) response to several immunological activators, including IL-1,70 are highly susceptible to a wide variety of experimental autoimmune disorders.
There is preliminary evidence that a defective HPA axis is present in both murine and human lupus. A significantly lower increase in plasma corticosterone concentrations upon stimulation by recombinant IL-1 is seen in lupus prone (MRL/lpr) mice.71 Aging of MRL/lpr mice, which is accompanied by an increase in autoantibody production, is associated with a decrease in hypothalamic CRH mRNA expression.72 Studies on the function of the HPA axis in patients with SLE are limited and often confounded by the effect of concomitant glucocorticoid treatment. A study on a group of active untreated female patients with SLE reports that the cortisol response to induced hypoglycaemia is significantly lower in patients than in healthy controls, indicating that some degree of HPA axis dysfunction does exist.73 The dysregulated HPA axis in SLE may be responsible for disease susceptibility and progression.
Table 3 summarises the role of hormones in the pathogenesis of SLE. Hormones may not have a direct causative role in SLE, but a milieu consisting of different values of hypothalamo–pituitary and gonadal hormones may create an endogenous environment for susceptible individuals to develop the disease. Changes in the concentrations of sex steroids, coupled with certain yet undiscovered environmental factors, may lead to disease flares and serve to explain the “wax and wane” nature of the disease.
Role of hormones in human SLE
IMMUNOPATHOLOGY
The basic pathological features of SLE are that of inflammation and blood vessel abnormalities, which include band or occlusive vasculopathy, vasculitis, and immune complex deposition. The best characterised organ pathology is in the kidney. By light and immunofluorescence microscopy, renal biopsies in patients with SLE display mesangial cell proliferation, inflammation, basement membrane abnormalities, and immune complex deposition, comprising immunoglobulins and complement components. On electron microscopy, these deposits can be visualised in the mesangium and the subendothelial or subepithelial surface of the basement membrane.
Other organ systems affected by SLE usually display non-specific inflammation or vascular abnormalities, although pathological findings are sometimes minimal. One example is cortical microinfarcts and bland vasculopathy with degenerative or proliferative changes in patients with central nervous system disease. Inflammation and necrotising vasculitis can rarely be found. Occlusive vasculopathy is a common histological feature associated with the presence of antiphospholipid antibodies. Atherosclerosis and tissue damage caused by hypertension, corticosteroids, and other drugs can be demonstrated in patients with long standing SLE.
Autoantibodies
The central immunological disturbance in patients with SLE is autoantibody production. These antibodies are directed at several self molecules found in the nucleus, cytoplasm, and cell surface, in addition to soluble molecules such as IgG and coagulation factors. Antinuclear antibodies are most characteristic and present in more than 95% of patients. Anti-double stranded DNA (ds-DNA) and anti-Sm antibodies are unique to patients with SLE. In fact, their presence is included in the classification criteria of SLE.74 The Sm antigen is designated as a small nuclear ribonucleoprotein (snRNP) and is composed of a unique set of uridine rich RNA molecules bound to a common group of core proteins and other proteins associated with the RNA molecules. Anti-Sm antibodies react with snRNP core proteins, whereas anti-DNA antibodies bind to a conserved nucleic acid determinant widely present on DNA. Anti-DNA antibody titres frequently vary over time and disease activity but anti-Sm antibody titres are usually constant.
The most remarkable feature of anti-DNA antibodies is their association with glomerulonephritis. Anti-DNA antibodies can be isolated in an enriched form from glomerular eluates of patients with active lupus nephritis and anti-DNA antibodies can induce nephritis in normal and severe combined immunodeficient mice.75,76 However, the correlation between anti-DNA antibodies and lupus nephritis in not complete because some patients with active nephritis are negative for anti-DNA antibodies, whereas some patients with persistent high titres of anti-DNA may not show renal involvement.
“The most remarkable feature of anti-DNA antibodies is their association with glomerulonephritis”
Anti-DNA antibodies differ in their properties, including isotype, ability to fix complement, and capacity to bind to the glomeruli causing pathogenicity.75 Only certain types of anti-DNA antibodies are pathogenic.77 The involvement of anti-DNA antibodies in lupus nephritis is suggested by several pieces of circumstantial evidence. First, clinical observations in most patients indicate that active nephritis is associated with raised anti-DNA titres and reduced total haemolytic complement values.78 Second, anti-DNA antibodies show preferential deposition in the kidneys, suggesting that DNA–anti-DNA antibody immune complexes are the main mediators of inflammation.79 Although renal injury may result from immune complexes containing anti-DNA antibodies, circulating immune complexes have been difficult to characterise because of their low concentration in serum. The formation of immune complexes in situ, instead of within the circulation, may be another possibility. Anti-DNA antibodies may bind to pieces of DNA adherent to the glomerular basement membrane via the histones or interact with additional glomerular antigens, such as C1q, nucleosomes, heparan sulfate, and laminin.80 The binding of anti-DNA antibodies to these antigens may initiate local inflammation and complement activation, and may also anchor immune complexes to the kidney sites, whether or not they are formed in the circulation or in situ.
Although an association between certain clinical features of SLE and autoantibodies such as anti-ribosomal P antibodies and psychosis, and anti-Ro antibodies and congenital heart block and subacute cutaneous lupus, has been well documented, the pathogenicity of these antibodies has not been adequately studied. The exact immunological mechanisms for injury remain to be elucidated. The pathogenesis of manifestations other than glomerulonephritis is less well understood, although immune complex deposition with activation of complement at relevant sites is a probable mechanism. This is demonstrated by the frequent association of hypocomplementaemia and signs of vasculitis at the sites of active SLE. Direct antibody mediated damage and cell mediated cytotoxicity on target tissues are other possible mechanisms.
Disturbances of the immune response
SLE is characterised by a myriad of immune system aberrations that involve B cells, T cells, and cells of the monocytic lineage, resulting in polyclonal B cell activation, increased numbers of antibody producing cells, hypergammaglobulinaemia, autoantibody production, and immune complex formation. It appears that excessive and uncontrolled T cell help in the differentiation and activation of autoantibody forming B cells is probably a final common pathway.
The activation of B and T cells requires stimulation by specific antigens. Irritating chemicals such as pristine, bacterial DNA and cell wall phospholipids, and viral antigens can induce anti-DNA antibodies in mice.77 Moreover, self antigens, such as DNA–protein and RNA–protein complexes may induce autoantibody production.81 Environmental antigens and self antigens are taken up by professional antigen presenting cells (APCs) or bind to induced antibodies on the surface of B cells. Both professional APCs and B cells process the antigens into peptides and present them to T cells through their surface HLA molecules. The activated T cells in turn stimulate the B cells to produce pathogenic autoantibodies. In addition to contact stimulation, the interaction of B and T cells is facilitated by several cytokines, such as IL-10, and requires accessory molecules such as those of the CD40/CD40L and B7/CD28/CTLA-4 systems to initiate a second signal.
“The number of B cells at all stages of activation is increased in the peripheral blood of patients with active systemic lupus erythematosus”
B cell activation is abnormal in patients with SLE. The number of B cells at all stages of activation is increased in the peripheral blood of patients with active SLE.82 These B cell abnormalities can precede the development of SLE. Activated lupus B cells have higher intracytoplasmic calcium responses than controls.83 There is also evidence that B cells in patients with SLE are more sensitive to the stimulatory effects of cytokines such as IL-6 than non-SLE B cells.84 Moreover, the phenomenon of epitope spreading has been demonstrated in both human and murine SLE.85 Thus, it appears that B cells in patients with SLE are more prone to polyclonal activation by antigens, cytokines, and other stimuli.
Abnormalities in T cell function are also evident in patients with SLE. The total number of peripheral blood T cells is usually reduced, probably because of the effects of anti-lymphocyte antibodies.86 There is a skewing of T cell function towards B cell help, leading to enhanced antibody production.87 Experiments have shown that the early events of T cell activation are defective in patients with SLE compared with controls.88,89 Although peripheral lupus T cells are activated, both their capacity for proliferation in response to mitogenic stimulation and IL-2 production are reduced.90–93 The reasons for the defective Th1 responses in SLE remain speculative. Downregulation by excessive Th2 cytokines, defective interaction between APCs and T cells, the suppressive effects of CD8+ T cells and natural killer (NK) cells, the presence of IL-2 inhibitors, and the downregulation of IL-2 receptors are possible mechanisms.94,95
Cytokine network in SLE
Cytokine profiles in patients with SLE have been studied extensively and are summarised in table 4. Peripheral blood mononuclear cells (PBMCs) from patients with SLE proliferate less than controls when stimulated with various antigens and mitogens.90 Supernatants from phytohaemagglutinin (PHA) or autologous mixed lymphocyte reaction stimulated lupus T cells produce less IL-2 than control T cells. Lupus T cells are less responsive to IL-2 stimulation than normal T cells.91–93 However, the expression of IL-2 in freshly prepared SLE PBMCs was increased compared with control PBMCs.96 Lupus T cells are capable of producing normal amounts of IL-2 in response to optimal stimulation with PHA combined with phorbol esters or with anti-CD28 antibodies.94,97 As mentioned previously, the impaired in vitro IL-2 production from lupus T cells is probably the result of many factors, one of which is the downregulating effects of certain Th2 cytokines.
Cytokines in patients with SLE
The recent discovery of the role played by IL-10 in the pathogenesis of SLE supports this hypothesis. IL-10 is a Th2 cytokine that acts as a potent stimulator of B cell proliferation and differentiation, and thereby a potential mediator of polyclonal B cell activation in SLE. Indeed, recent studies have shown that spontaneous production of IL-10 from SLE peripheral blood B cells and monocytes is significantly higher than that of controls.98,99 The expression of IL-10 transcripts is significantly increased in the non-T cell population of PBMCs from patients with SLE compared with controls.100 Moreover, serum IL-10 concentrations are higher in patients with SLE than in controls and are correlated with clinical and serological disease activity, and anti-DNA antibody titres.101–103 Furthermore, an increased ratio of IL-10 to interferon γ secreting cells in the PBMCs of patients with SLE correlates with disease activity.104
The increase in IL-10 production may be a cause of the defective in vitro Th1 response in lupus T cells. This is suggested by a study demonstrating that the addition of blocking antibodies to IL-10 significantly enhanced the proliferative response of lupus PBMCs.105 Conversely, PBMCs from patients with SLE and inactive disease cultured with IL-10 showed a significant augmentation of anti-DNA antibody production.106
IL-12, a heterodimeric cytokine produced by B cells, macrophages, and dendritic cells, promotes cell mediated immune responses but exerts some inhibitory activities on humoral responses.107 IL-12 production was found to be impaired in stimulated PBMCs from patients with SLE compared with matched controls.108,109 The defect in IL-12 production probably lies in the monocytes but not the B cells.110 In contrast, the addition of IL-12 to lupus PBMCs significantly inhibits both spontaneous and IL-10 stimulated immunoglobulin and anti-DNA antibody production.111 Moreover, the production of anti-DNA antibodies by PBMCs from patients with SLE and active disease is inhibited by culturing the cells with IL-12.106 The results from these studies suggest that dysregulation of the IL-10–IL-12 balance plays a crucial role in the impaired cellular immune responses seen in patients with SLE.
Defective immune regulation
The clearing of immune complexes by phagocytic cells is defective in patients with SLE.7 This results partly from the reduced numbers of CR1 receptors for complement and functional defects of the receptors on cell surfaces.112,113 Defective clearance may also result from inadequate phagocytosis of IgG2 and IgG3 containing complexes. Allelic polymorphisms of the IgG receptors (FcγR) have recently been described. Some of the polymorphic alleles (FcγRIIA and FcγRIIIA) are associated with lower binding of the Fc portions of IgG2 and IgG3, and hence impaired clearance of immune complexes.7,114 Indeed, the FcγRIIA and FcγRIIIA genotypes have been associated with susceptibility to SLE and nephritis in certain ethnic groups.7–9,115 Although consistent results cannot be obtained in patients of different ethnicities, impaired immune complex clearance by phagocytes is an important pathogenetic mechanism in SLE.
A recent study also demonstrated that non-inflammatory engulfment phagocytosis of apoptotic cells is impaired in patients with SLE.116 Persistently circulating apoptotic waste may serve as an immunogen for the induction of autoreactive lymphocytes and as an antigen for immune complex formation.
“Impaired immune complex clearance by phagocytes is an important pathogenetic mechanism in systemic lupus erythematosus”
The synthesis and secretion of pathogenic autoantibodies in SLE is driven by the interaction of CD4+ and CD8+ helper T cells, and double negative T cells (CD4− CD8−) with B cells.117 Therefore, cells that normally suppress B cell activation, such as CD8+ suppressor T cells and NK cells are defective in their suppressive activity. It has been shown that CD8+ T cells and NK cells from patients with active SLE are often incapable of downregulating polyclonal immunoglobulin synthesis and autoantibody production.87 One recent study reported that CD8+ T suppressor cell function was impaired in patients with active SLE.118 This impaired suppression of B cells may be one factor that leads to the perpetuation of the disease.
In normal healthy subjects, overproduction of antibodies is prevented by an idiotype network. This network is probably defective in patients with SLE, leading to dysregulation of autoantibody production.119
Table 5 summarises the abnormalities in the immune response and immunoregulation in patients with SLE.
Summary of abnormal immune responses and immunoregulation in patients with SLE
APOPTOSIS AND SLE
Apoptosis, or programmed cell death, is a process that leads to the ordered destruction of cells, avoiding the release of intracellular contents into the extracellular microenvironment, where they have a powerful inflammatory effect. Defective apoptosis leading to the prolonged survival of pathogenic lymphocytes was thought to be one disease mechanism for SLE. This hypothesis was supported by observations in murine lupus models. MRL/lpr mice are characterised by the presence of the lpr gene, which is associated with defective Fas (CD95) receptors on the surface of lymphocytes. The interaction of Fas and Fas ligand (FasL) transduces an active signal for cellular apoptosis.120 Defective Fas mediated apoptosis in MRL/lpr mice results in massive lymphoproliferation and the development of a severe lupus-like disease with immune glomerulonephritis.121 Gld/gld mice, characterised by a mutation in the FasL gene leading to a non-functional FasL molecule, also develop lymphoproliferation, hypergammaglobulinaemia, and immunoglobulin deposits in the kidneys.122
However, there are several problems with this model. First, the massive lymphoproliferation resulting from defective apoptosis seen in the MRL/lpr and Gld/gld mice is not characteristic of human SLE, which conversely is often associated with profound lymphopenia. Second, the expression of both Fas and FasL is normal in patients with SLE.123,124 Lastly, apoptosis of peripheral lymphocytes in patients with SLE has been shown to be increased compared with controls.125,126
The increased rate of apoptosis in SLE would theoretically increase the chance of leakage of intracellular antigens that may either trigger an autoimmune response or participate in the formation of immune complexes. Under normal circumstances, apoptotic cells are engulfed by macrophages in the early phase of apoptotic cell death without inducing inflammation or the immune response. Recent studies have shown that the clearance of apoptotic cells by macrophages in patients with SLE is impaired.116 This is not confined to monocytes and macrophages of the peripheral blood, but also occurs in the germinal centres of lymph nodes.127,128
The reasons for the defective clearance of apoptotic cells in SLE are not clear. It could be the result of quantitative or qualitative defects of the early complement proteins, such as C2, C4, or C1q. Patients with homozygous deficiencies in these complement components develop a severe lupus-like disease early in life.129 The C1q receptors on the surface of phagocytes constitute an extremely important mechanism for the clearance of apoptotic cells.130 Patients or mice with homozygous C1q deficiency develop autoantibodies and a lupus-like syndrome apparently because of the inability to eliminate apoptotic cells effectively, which leads to an increase in the exposure of antigens to the immune system.129,131 Mice with a targeted deletion of C1q show glomerulonephritis with deposits of immune complexes and apoptotic cells in the glomeruli.131
Anti-C1q antibodies can be found in a large proportion of patients, particularly those with renal disease.132 This may result in a functional deficiency of the receptor protein. It is unlikely that anti-C1q antibodies represent the primary abnormality in most patients with SLE, but they would certainly provide a mechanism for persistence of the disease and flares.133
Although bcl2 expression is increased in some lupus T cells, the overall rate of lymphocyte apoptosis is increased in SLE. Taken together with the impaired clearance mechanisms for these apoptotic materials seen in patients with SLE, this may predispose individuals to the development of antibodies against nucleosomes, which contain materials such as histones and dsDNA.
ENVIRONMENTAL TRIGGERS
Although genetic factors and the hormonal milieu may create a predisposition towards SLE, the initiation of the disease probably results from several environmental triggers and exogenous factors (table 6). Infectious agents may induce specific responses by molecular mimicry and disturb immunoregulation; diet affects the production of inflammatory mediators; toxins/drugs modify cellular responsiveness and immunogenicity of self antigens; and physical/chemical agents, such as ultraviolet (UV) light, can cause inflammation, induce cellular apoptosis, and cause tissue damage. The impingement of these environmental triggers on predisposed individuals is probably highly variable and could be a further explanation for disease heterogeneity, in addition to its alternating periods of flares and remission.
Environmental factors that may be relevant in the pathogenesis of systemic lupus erythematosus
Chemical/physical factors
Many drugs such as procainamide and hydralazine, which are aromatic amines or hydrazines, can induce a lupus-like syndrome, especially in individuals who are genetically slow acetylators.134 Both aromatic amines and hydrazines, and their derivatives, can be found in a wide variety of compounds used in agriculture and industry, and commercial applications. Hydrazine itself also occurs naturally in tobacco and tobacco smoke. Lupus-like syndromes have been reported in individuals who have ingested or have been in contact with these agents.135
Permanent hair colouring solutions contain aromatic amines that can be absorbed through the scalp. Chronic use of hair dyes has been associated with the development of SLE.136 However, subsequent studies did not confirm such an association.135,137,138 A recent study reported only a weak association between the risk of SLE and permanent hair dyes or smoking.139
Take home messages
-
The patho-aetiology of systemic lupus erythematosus (SLE) probably involves complicated and multifactorial interactions among various genetic and environmental factors
-
Multiple genes contribute to disease susceptibility, including genes encoding complement and other components of the immune response, in addition to major histocompatibility complex class I and II genes
-
The interaction of sex, hormonal milieu (particularly oestrogenic versus androgenic activity), and the hypothalamo–pituitary–adrenal axis modifies this susceptibility and the clinical expression of the disease
-
Defective immune regulatory mechanisms, such as the clearance of apoptotic cells and immune complexes, are important contributors to the development of SLE
-
The loss of immune tolerance, increased antigenic load, excess T cell help, defective B cell suppression, and the shifting of T helper 1 (Th1) to Th2 immune responses leads to B cell hyperactivity and the production of pathogenic autoantibodies
-
In addition, environmental factors, such as chemicals and drugs, ultraviolet light, dietary factors, viruses, and environmental oestrogen are probably required to precipitate the onset of the disease
Exposure to sunlight is a well known environmental factor in the induction and exacerbation of both cutaneous and systemic lupus erythematosus. UV light, especially UVB, is an important trigger in many patients with SLE. There is good evidence that exposure of skin to UV light alters the location and/or chemistry of DNA, in addition to Ro and nRNP antigens, and probably enhances their immunogenicity.140 Recent studies have demonstrated that UV light induces the apoptosis of human keratinocytes, leading to the formation of clusters or blebs on the surface of dying cells, which contain both nuclear and cytoplasmic antigens.141,142 This provides a mechanism for the exposure of self antigens to the immune system and provokes autoimmunity.
Dietary factors and infectious agents
Although several dietary factors and infectious agents are implicated in the pathogenesis of SLE, none of these has been consistently demonstrated in more than one study. The ingestion of alfalfa sprouts that contain L-canavanine has been linked to the development of lupus-like symptoms in several case reports.143 Infectious agents such as viruses might theoretically initiate or cause a flare in SLE by activating B cells, damaging tissues to release autoantigens, and triggering the disease by molecular mimicry. However, viral “footprints” have not been consistently demonstrated in the tissues of patients with SLE.144 Thus, there is little evidence to support that one infectious agent causes SLE, although this remains a possibility and warrants further investigations.
Environmental oestrogens
The exposure of humans to environmental oestrogens is believed to increase over the years through the consumption of meat and milk products of livestock that are fed with synthetic oestrogens.145,146 Moreover, oestrogens are increasingly used by postmenopausal women and for contraception. Chronic oestrogen exposure in prepubertal non-immune mice has been shown to influence thymic development and hence immune tolerance.145 It is therefore plausible that exposure to oestrogenic compounds during fetal life could be a potential immunological hazard. In fact, prenatal diethylstilbesterol exposure has been linked to autoimmune disorders, although further confirmation is needed.147 HRT and the use of OC pills have also been associated with a small increase in the risk of SLE development.37,38 Thus, environmental oestrogens and endocrine disrupting chemicals may be important triggers for autoimmunity in susceptible individuals. The ultimate development of the disease will depend upon several factors, which include genetic background, sex, age, dose and duration of exposure, and immune status at the time of contact with these agents.
SUMMARY
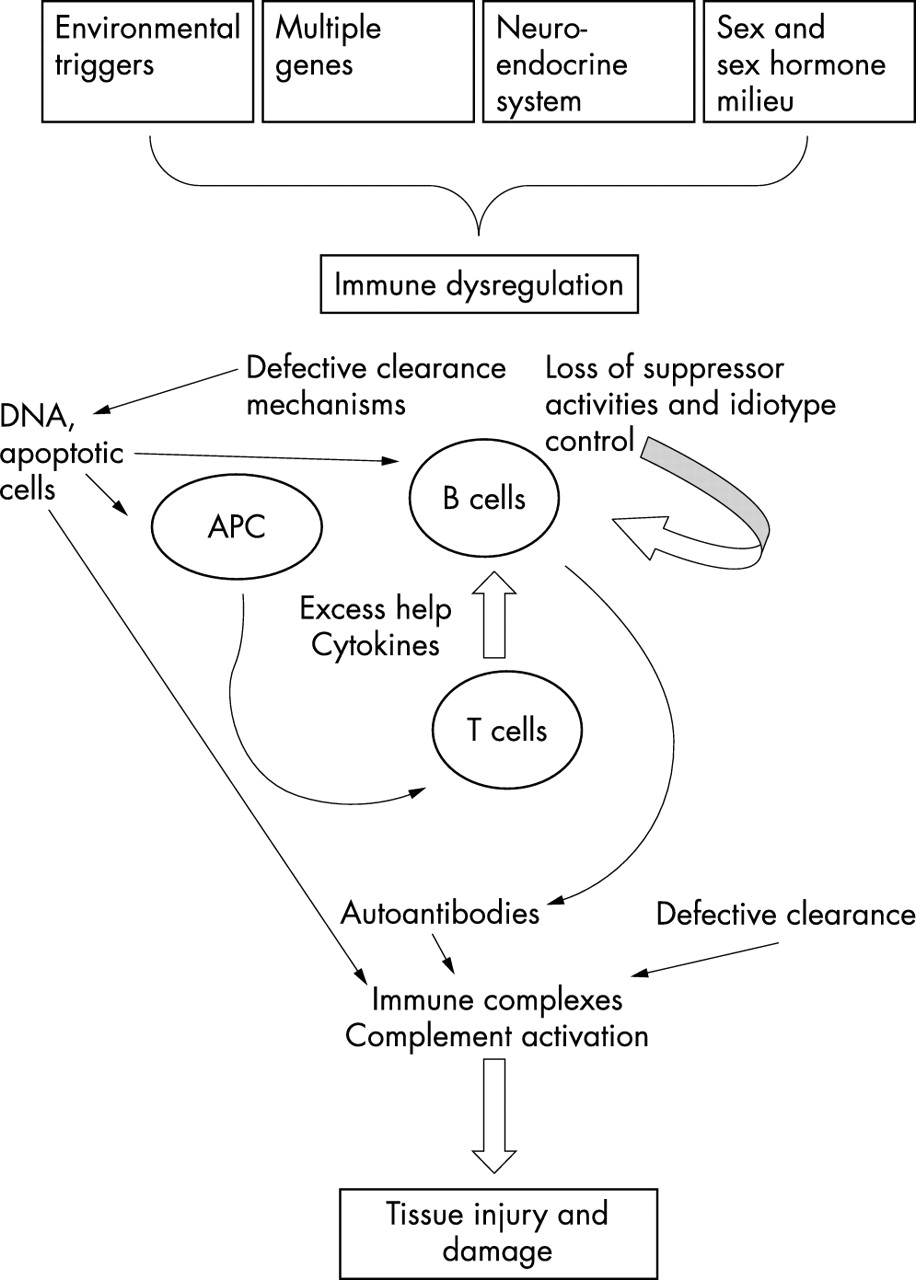
The pathogenesis of SLE is summarised diagrammatically in fig 1. Multiple genes confer susceptibility to disease development. Interaction of sex, hormonal milieu, the HPA axis, and defective immune regulation, such as clearance of apoptotic cells and immune complexes, modify this susceptibility. The loss of immune tolerance, increased antigenic load, excess T cell help, defective B cell suppression, and shifting of Th1 to Th2 immune responses lead to cytokine imbalance, B cell hyperactivity, and the production of pathogenic autoantibodies. Finally, certain environmental factors are probably needed to precipitate the onset of the disease.
{kind=link}
The pathogenesis of systemic lupus erythematosus. APC, antigen presenting cell.