

Abstract
Psoriasis vulgaris (PsV) and psoriatic arthritis (PsA) are interrelated disorders, PsA representing a disease within a disease. From an epidemiological perspective, the genetic contributions of PsV and PsA are now well documented. HLA-C is firmly established as a PsV/PsA gene, with HLA-Cw*0602 as a major risk allele. Fine-mapping studies within the MHC region in PsV and PsA have identified novel loci that are independent of the HLA-Cw6 allele. Recent genome-wide association scans have led to a substantial increase in the number of candidate genes reaching genome-wide significance in PsV and PsA cohorts. Most of these genes can be grouped into an integrated pathogenic model of PsV/psoriatic disease comprising distinct signaling networks affecting skin barrier function (LCE3, DEFB4, GJB2), innate immune responses involving nuclear factor-κB and interferon signaling (TNFAIP3, TNIP1, NFKBIA, REL, FBXL19, TYK2, NOS2), and adaptive immune responses involving CD8 T lymphocytes and interleukin 23 (IL-23)/IL-17-mediated lymphocyte signaling (HLA-C, IL12B, IL23R, IL23A, TRAF3IP2, ERAP1). Further development of a global genetic risk score and inclusion of potential gene/gene and gene/environment interactions will likely enhance the predictive value of recently identified genetic variants.
Genetics of Psoriasis Vulgaris and Psoriatic Arthritis
The most dominant genetic effect of psoriasis vulgaris (PsV)/psoriatic disease exists within the major histocompatibility complex (MHC) region located on chromosome 6p21.3. This region was initially localized to ∼300 kb segment known as PSORS1. Based on a resequencing study by Nair, et al human leukocyte antigen (HLA)-Cw*0602 was revealed to be the PSORS1 risk variant that confers susceptibility to PsV1. The association of PsV with several HLA-B alleles and non-HLA genes (such as CDSN, HCR, and PSORS1C3) within the MHC has also been reported, but their independence from HLA-C has not been established2. Recently, 2 other loci associated with PsV susceptibility that are independent of HLACw*0602 were identified: single-nucleotide polymorphism (SNP) rs2073048, a polymorphism within c6orf10 gene; and SNP rs13437088, a variant 30 kb centromeric of HLA-B and 16 kb telomeric of MICA3.
HLA-Cw*0602 is also associated with psoriatic arthritis (PsA); however, the magnitude of association is lower than in PsV2. HLA antigens associated with PsA include HLA-B13, HLA-B27, HLA-B38/39, HLA-B57, and HLA-DRB1*042. Non-HLA candidates include TNF-α promoter polymorphisms (TNF −238G/A and −857T) and MIC alleles, of which the trinucleotide repeat polymorphism MICA-A9 that corresponds to MICA*002 appears to be associated with PsA independent of HLA-Cw6, MICB, or TNF2. However, a more recent study concluded that no MIC alleles were associated with PsA after adjusting for known HLA alleles4. Recent fine-mapping of the MHC region in PsA noted a significant association with SNP rs1150735, located 1.5 kb upstream from ring finger protein 39 gene (RNF39)5.
Multiple genome-wide linkage studies and candidate-gene studies have been performed in an attempt to identify non-MHC candidate genes in PsV, but very few genes have been consistently replicated6,7.
Genome-wide association scans (GWAS) represent an important advancement for gene identification in PsV/psoriatic disease. Presently 6 GWAS have been published involving either PsV or PsA cohorts, 5 of which are cohorts of European ancestry totaling 5335 patients with PsV8,9,10,11,12. The presence of inflammatory arthritis was not systematically evaluated in all patients with PsV; however, at least 21% of the patients were documented to have PsA. Nineteen loci have reached genome-wide significance among whites (Figure 1).
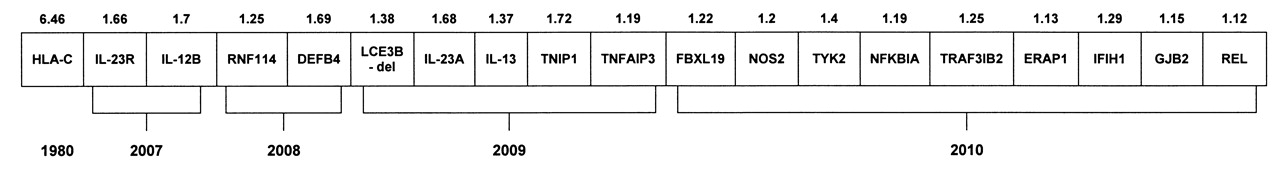
Candidate genes reaching genome-wide significance in Caucasian psoriasis patients. The magnitude of the risk for each gene is shown above each gene, and the year identified is shown below.
GWAS has identified many candidate genes that strongly suggest that the pathophysiology underpinning PsV and PsA reflect an integrated complex interplay encompassing distinct signaling networks in skin barrier function and innate immune responses8. Genes implicated in skin barrier function include LCE3, DEFB4, and GJB2. A genome-wide investigation targeting copy number variations identified a 32.2-kb deletion within the PSORS4 locus that encompasses both LCE3B and LCE3C genes with increased susceptibility to PsV13. Increased DEFB4 copy number is associated with PsV, with each additional copy above 2 copies increasing the relative risk, and a variant residing in GJB2 is a PsV susceptibility locus14,15. Variations within numerous genes that encode proteins critical for nuclear factor-κB (NF-κB) signaling and subsequent transcription have reached genome-wide significance (TNFAIP3, TNIP1, NFκBIA, REL, TYK2, IFIH1, and FBXL19). Finally, genes in the adaptive immune response involving CD8 T lymphocytes and Th-17 lymphocyte signaling (HLA-C, ERAP1, IL12B, IL23R, IL23A, and TRAF3IP2) have also reached genome-wide significance.
TRAF3IP2 is of particular interest due to its biological relevance and the suggestion that its genetic contribution may be more prominent in PsA than PsV. TRAF3IP2 encodes Act1, an adapter protein essential for Th-17-mediated inflammatory responses, including IL-17-dependent NF-κB activation16. In epithelial cells, IL-17-dependent receptor ligation recruits Act1 to bind to the cytoplasmic tail of the IL-17 receptor17. This results in incorporation of TRAF6 (and possibly TRAF3) into the signaling complex and the subsequent downstream activation of the NF-κB, p38, and MAPK pathways16,17. It is reasonable to assume that genetic variation within TRAF3IP2 and the resultant dysregulation of Act1 may significantly alter IL-17 signaling and subsequent activation of NF-κB pathways12. Studies in TRAF3IP2 knockout mice also suggest that Act1 is a negative regulator of humoral immunity through its inhibitory effect on CD40 and BAFFR-mediated signaling12.
PsA-weighted Genes from GWAS
Multiple genes achieving genome-wide significance are shared between PsV and PsA cohorts, which strongly reiterates the interrelatedness of these inflammatory diseases. With regard to PsA-weighted genes, there is a slightly higher effect size for TRAF3IP2 in PsA (p = 4.5 × 10−12) compared with psoriasis (p = 2.0 × 10−6)11. Also, a variant residing in FBXL19 is more frequent in PsA than PsV as evidenced by a minor allele frequency of 0.412 in SNP rs10782001 in PsA compared with 0.385 in PsV (p = 0.02)11.
Predicting Disease Risk from Genetic Variants
With the exception of HLA-Cw6 (and marker HLA-Cw0602), the effect size for each individual gene identified in PsV and PsA is small and has minimal predictive ability. One way to improve the predictive capacity is to combine multiple loci of mild to modest effects into a global genetic risk score (GRS), as done recently by Chen, et al18. They found that a GRS combining 10 psoriasis risk loci identified significantly more risk than individual SNP markers. Further, a weighted GRS that accounts for the odds ratio of each allele is a better discriminator than just a simple count of the disease alleles among cases and controls. Finally, further stratifying the highest quartile of the weighted GRS increased the risk of psoriasis 10-fold compared with persons in the lowest quartile.
Once genes are identified, further genetic complexities may need to be evaluated. Recently, the first compelling statistical interaction was noted between 2 GWAS loci, SNP tagging HLA-Cw*0602 and ERAP114. This finding is of particular interest given the role of HLA-C and ERAP1 in class I antigen presentation.
Predicting Disease Prognosis from Genetic Variants
Phenotypic association with disease expression for PsV has been most strongly noted for HLA-Cw*0602. As summarized by Duffin, et al, HLA-Cw*0602 has been associated with early age of onset of psoriasis, higher prevalence of family history, presence of guttate psoriasis, and Koebner phenomenon2. For PsA, HLA-B39 alone, HLA-B27 (only in the presence of HLA-DR7), and HLA-DQ3 (only in the absence of HLA-DR7) conferred an increased risk for disease progression along with HLA-DRB1*04 allele, which is associated with radiographic progression19. PsA patients carrying both HLA-Cw6 and HLA-DRB1*07 alleles have a less severe course of arthritis20. A recent study has identified 2 IL13 SNP (rs20541 and rs1800925) that were highly associated with susceptibility to PsA but not to PsV21. Finally, the AA genotype of the IL4R SNP (rs1805010, encoding the amino acid change I50V) is associated with joint erosions in PsA13. In patients with no radiographic joint damage, the carrier frequency for the AA genotype was 16%, compared with 27% among those with 1 to 5 damaged joints and 40% among those with ≥ 5 damaged joints22. The estimated increase in the rate of damaged joints among patients with the AA genotype was 53%. Of note, the effect sizes for most of these associations are modest, and their clinical utility and discriminating ability have yet to be determined.
Despite the recent advances in GWAS studies, a substantial portion of the genetic risk for PsV and in particular PsA (given the high sibling heritability) remains to be identified. However, because about 90% of patients with PsA develop skin lesions prior to the onset of joint disease23, and because only 1% to 2% of European-origin populations develop psoriasis, the predictive value of genetic testing among patients with newly diagnosed cutaneous psoriasis is greater than can be expected in the general population. Further GWAS studies are warranted, particularly in PsA cohorts, and next-generation sequencing technologies and systematic evaluation of structural variants must be conducted to identify novel causal variants that likely account for a significant proportion of the missing heritability in PsV and PsA.








{kind=link}