

Abstract
Objective. Clinical markers are needed to identify scleroderma patients at risk for pulmonary arterial hypertension (PAH) since early therapy may improve survival. We investigated whether increased numbers of telangiectases in scleroderma associate with measures of pulmonary vascular disease.
Methods. One hundred forty-seven consecutive adult patients with scleroderma were enrolled in this cross-sectional study and scored for the presence of matted telangiectases on 11 body areas. Per body area, telangiectases were scored as 0 if none were present, 1 if there were fewer than 10 telangiectases, and 2 if 10 or more telangiectases were counted. Linear regression analysis was performed to assess the association between right ventricular systolic pressure (RVSP) and telangiectasia score, adjusted for age, race, smoking status, scleroderma subtype, disease duration, and autoantibody status. Logistic regression analysis was performed with PAH by right-heart catheterization (RHC) as the dependent variable.
Results. The mean telangiectasia score was 6.0 (SD 4.5, range 0–20). RVSP and telangiectasia score were positively correlated (r = 0.271, p = 0.001). The mean RVSP increased by 10.9 mm Hg for every 10-point increase in telangiectasia score (95% CI 3.6–18.3 mm Hg, p = 0.004), adjusted for potential confounders. The adjusted relative odds of PAH by RHC were 12.4 for patients with a 10-point increase in telangiectasia score (95% CI 1.78–85.9, p = 0.01).
Conclusion. Increased numbers of telangiectases strongly associate with the presence of pulmonary vascular disease. Telangiectases may be a clinical marker of more widespread aberrant microvascular disease in scleroderma.
Telangiectases are common manifestations of microvascular changes in scleroderma. They are vascular lesions composed of vasodilated post-capillary venules without evidence of neovascularization or inflammation1,2. Telangiectases develop primarily on the fingers, hands, face, and mucous membranes, but may also be found on the limbs and trunk. Although they are thought to occur more commonly in patients with limited scleroderma, telangiectases become more numerous over time in both limited and diffuse subtypes. The biological mechanism causing the development of telangiectases in scleroderma is unknown. They may be the result of an aberrant attempt at increasing blood perfusion to hypoxic tissues that is a consequence of the loss of normal circulation in affected tissues. Thus, telangiectases may be markers of ongoing vascular injury and failed vascular repair that is thought to occur in multiple vital organs in scleroderma3.
We hypothesize that the presence of cutaneous telangiectasia is a sign of an ongoing vascular insult that could serve as an easily accessible clinical biomarker for systemic vascular defects, including pulmonary vascular disease that may progress to pulmonary arterial hypertension (PAH). In this cross-sectional study, we evaluated whether increased numbers of telangiectases associate with evidence of PAH in patients with scleroderma.
MATERIALS AND METHODS
Consecutive, consenting subjects with scleroderma were enrolled during routine appointments to the Johns Hopkins Scleroderma Center from February to April of 2007 by one of 2 experienced scleroderma investigators. A predesigned telangiectasia scoring form was used for each evaluation, and the methodology for scoring each subject was agreed upon by all investigators prior to subject enrollment. The only inclusion criteria were the ability to obtain consent and a diagnosis of scleroderma defined as either the American College of Rheumatology criteria for scleroderma4, having at least 3 of 5 features of the CREST syndrome (calcinosis, Raynaud’s, esophageal dysmotility, sclerodactyly, telangiectases) or having definite Raynaud’s phenomenon, abnormal nailfold capillaries, and the presence of a scleroderma-specific autoantibody.
Subjects were scored for the presence of matted, nonstellate telangiectases (Figure 1). Body areas assessed included face, hands, arms, chest and abdomen, back, legs, and feet. For each body area, telangiectases were scored as 0 if no telangiectases were present, 1 if there were fewer than 10 telangiectases, and 2 if 10 or more telangiectases were counted. The total possible telangiectasia score was 22. To assess for interrater reliability of the scoring method, 20 additional patients were independently scored by both investigators during 1 clinic encounter, blinded to the other observer’s score.

Two different types of telangiectasia: (A) matted; (B) stellate.
Demographic data, disease duration, clinical disease characteristics, autoantibody status, and clinical outcome measures were extracted from previously recorded information in our database for all subjects. The date of first non-Raynaud’s symptom was used to calculate disease duration. Scleroderma type was defined in the method outlined by LeRoy, et al: limited with skin involvement that does not extend above the elbow or knee, and diffuse with skin disease proximal to elbows or knees or involving the trunk5. Severe Raynaud’s phenomenon was defined by a history of digital pitting scars, ulcers, or gangrene. Measures of cardiopulmonary disease included the echocardiogram and pulmonary function test (PFT) performed closest to the time of the study visit. Current medication use and the presence or absence of other comorbid conditions were determined during the study visit using these predetermined criteria: peripheral arterial disease (absent peripheral pulses, history of abnormal ankle-brachial indices, history of claudication, or history of amputation), coronary artery disease [a history of angina; abnormal electrocardiogram (ECG), standard exercise ECG test, exercise or pharmacologic stress imaging study, or coronary angiogram; or a history of coronary revascularization], atherosclerotic cerebrovascular disease (prior transient ischemic attack or thrombotic or embolic stroke), hypertension (systolic blood pressure > 140 mm Hg, diastolic blood pressure > 90 mm Hg, or antihypertensive medication use), and renal disease (renal insufficiency documented by abnormal creatinine). Assessments for PAH via right-heart catheterization (RHC) data were extracted from subjects’ clinical charts when available.
Echocardiograms
Right ventricular systolic pressure (RVSP) estimated by Doppler echocardiography was used as a surrogate for pulmonary vascular disease. RVSP ≥ 45 mm Hg was used as evidence of pulmonary vascular disease consistent with PAH as reported6,7. If an RVSP estimate was not recorded, all echocardiograms that lacked an adequate tricuspid jet and had no right ventricular hypertrophy were presumed to be normal, and a value of 30 mm Hg was imputed. An estimated RVSP was available in 119 patients and imputed in 28 patients. In 3 patients, an estimated RVSP could not be imputed, given insufficient data. Echocardiograms were performed at diverse clinical sites, and reports were reviewed by the investigators according to a predesigned protocol.
Pulmonary function tests (PFT)
The most recent spirometry, helium lung volumes, and carbon monoxide diffusing capacity (DLCO) results were recorded for each subject. PFT were performed at various sites and all measurements of forced vital capacity (FVC) and DLCO were standardized according to the US National Health and Nutrition Examination Survey and Knudson, et al, respectively8,9.
RHC
Among subjects who had RHC performed as part of routine clinical care, PAH was defined by a mean pulmonary artery pressure (MPAP) ≥ 25 mm Hg and a wedge pressure (PCWP) < 15 mm Hg. Pulmonary venous hypertension (PVH) was defined by MPAP ≥ 25 mm Hg and PCWP ≥ 15 mm Hg.
Statistical analyses
Exploratory data analysis for our explanatory variable of interest, total telangiectasia scores (0–22), and our primary outcome, RVSP, was performed including stem and leaf plots, box plots, and ladder of powers histograms. For some analyses, subjects were grouped into tertiles of telangiectasia score (0–3, 4–7, 8+). Differences in continuous and dichotomous or categorical clinical variables among the 3 groups were assessed by analysis of variance (ANOVA) and Fisher’s exact test, respectively. Interrater reliability was assessed by intraclass correlation coefficient.
The correlation between RVSP and telangiectasia score was assessed by Pearson’s correlation coefficient. In order to determine the strength of association of our clinical measure (telangiectases) as a predictor of pulmonary vascular disease, regression models were developed with RVSP as the dependent variable and telangiectasia score as a continuous explanatory variable. Simple linear regression analyses were then performed to assess bivariate associations between RVSP and telangiectasia scores, and between RVSP and other scleroderma characteristics. Multivariable linear regression analysis was performed to evaluate the relationship between RVSP and telangiectasia scores, adjusted for age, race, smoking status, scleroderma subtype, disease duration, and autoantibody status. These variables were fixed in all analyses given their clinical relevance. Regression diagnostics were performed, including Q-Q plots and the Shapiro-Wilk test for normality of studentized residuals. As RVSP was not normally distributed, the above analyses were also performed with logarithmically transformed RVSP. Bivariate and multivariable linear regression analyses with RVSP as the dependent variable and facial telangiectasia score alone (range 0–2) as a possible explanatory variable were also performed.
Secondary analyses were performed with the presence of PAH by RHC as the dependent variable. The Hosmer-Lemeshow test confirmed model goodness-of-fit. Differences in mean total telangiectasia score between patients with PAH, PVH, and normal RHC were explored by ANOVA with a Bonferroni correction.
Serum soluble endoglin levels tested by ELISA (R&D Systems Inc., Minneapolis, MN, USA) as part of another study were available in 11 patients. We assessed the correlation between endoglin levels and total telangiectasia scores by Pearson’s correlation coefficient. The correlation between severe Raynaud’s phenomenon and total telangiectasia score was also explored. Statistical significance was defined as a 2-sided p value ≤ 0.05. Statistical analyses were performed using Stata 10.0 (Stata Corp., College Station, TX, USA).
RESULTS
The study population consisted of 147 patients with scleroderma with a mean age of 54.3 years (SD 13.3 yrs). Most of the participants were women (86.4%), and 74.2% were white. Among all subjects, 59.2% had limited cutaneous disease and 40.8% had diffuse cutaneous scleroderma. The mean disease duration was 9.4 years, and the mean duration since Raynaud’s onset was 13.8 years. Comorbid vascular disease was seen in 27.2% of subjects. Of the study population, 21.9% had anticentromere antibodies (ACA), and 21.2% had scleroderma-70 antibodies. Subjects were frequently treated with calcium channel blockers (53.1%), angiotensin-converting enzyme inhibitors or angiotensin receptor blockers (36.7%), or antiplatelet medications (26.5%).
Telangiectasia score and its association with scleroderma characteristics
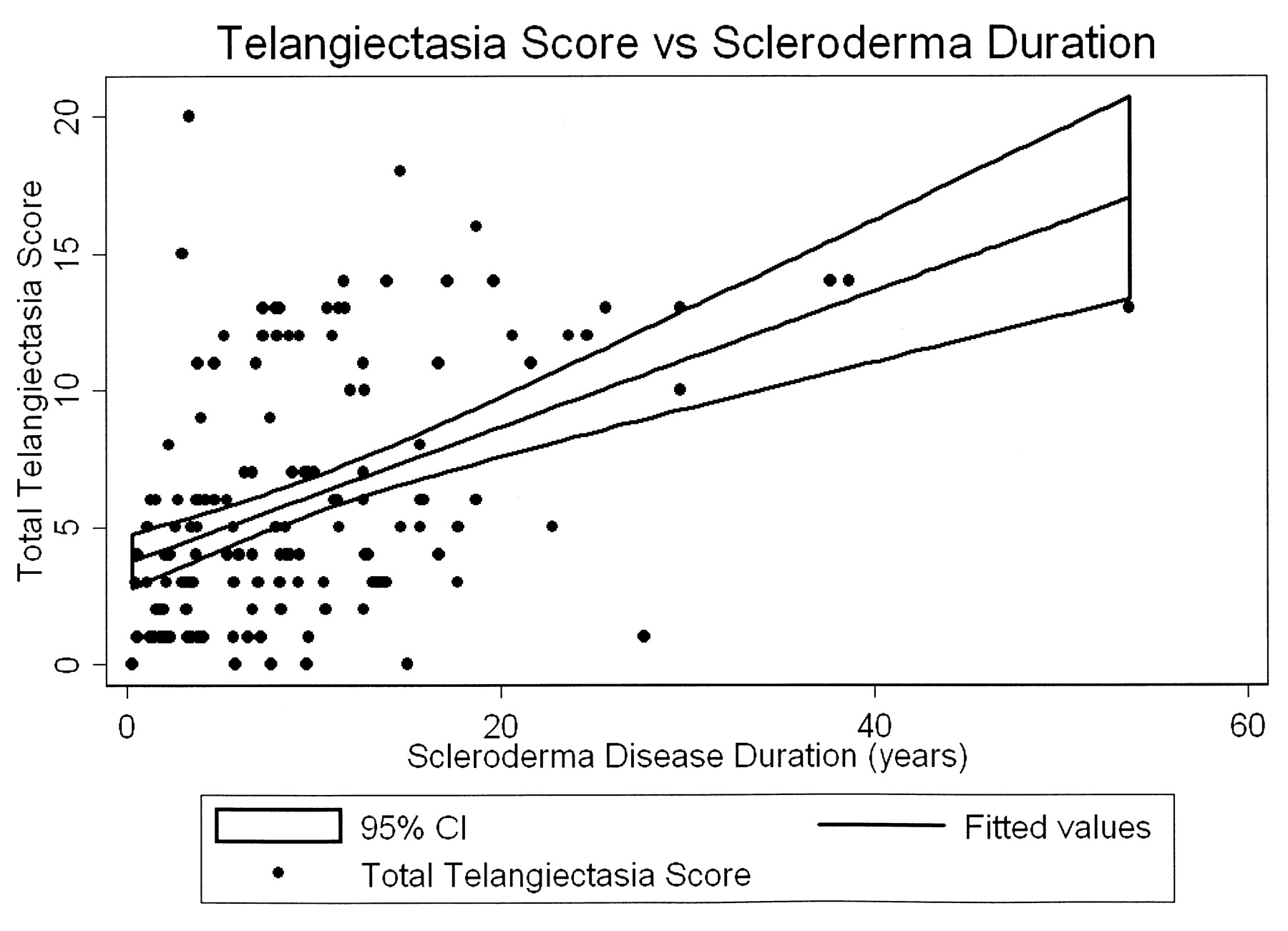
Telangiectasia assessment was highly consistent between both observers (interclass correlation coefficient = 0.95). Telangiectasia scores ranged from 0 to 20, and the mean telangiectasia score was 6.0 (SD 4.5). Fifty-four patients had a telangiectasia score of 0–3, 50 had a score of 4–7, and 43 had a score of 8–20. Baseline characteristics of our patients by tertile of telangiectasia score are detailed in Table 1. Patients in the second and third tertiles of telangiectasia score were older (p < 0.0001) and had a longer duration of scleroderma (p < 0.0001). African American patients had lower telangiectasia scores than their white counterparts (p = 0.007). More former and current smokers were present in second and third telangiectasia tertiles (p = 0.03). Patients in higher tertiles were also more commonly ACA-positive (p = 0.04) and more likely to be treated with phosphodiesterase-5 inhibitors (p < 0.001). Gender, scleroderma subtype, nonscleroderma vascular disease, renal disease, and anti-Scl70 and anti-RNP antibodies did not differ between tertiles of telangiectasia score. The strong relationship between disease duration and telangiectasia score is illustrated in Figure 2.

The mean telangiectasia score increased by 2.5 for every 10-year increase in scleroderma disease duration (p < 0.001).
Baseline characteristics of study participants.
Estimated RVSP and telangiectasia score
The mean RVSP was 39.6 mm Hg (SD 18.1 mm Hg), and 30 patients (20.8%) had an RVSP ≥ 45 mm Hg. The mean RVSP increased from 34.4 mm Hg in the lowest tertile to 48.1 mm Hg in the highest tertile of telangiectasia score (Table 2). RVSP and telangiectasia score were positively correlated (r = 0.271, p = 0.001). In bivariate analysis (Table 3), the mean RVSP increased by 10.8 mm Hg for a 10-point increase in total telangiectasia score (p = 0.001); this relationship is illustrated in Figure 3. Additional bivariate analyses demonstrated that the mean RVSP was also positively associated with age (p < 0.001), limited disease subtype (p = 0.047), and scleroderma disease duration (p = 0.07). Although the mean RVSP increased by 6.5 mm Hg comparing subjects with interstitial lung disease to those without, this was not statistically significantly different (p = 0.08), but there was a negative correlation with FVC and DLCO. In multivariable analysis, the mean RVSP increased by 10.9 mm Hg for every 10-point increase in telangiectasia score (p = 0.004), adjusted for age, race, disease duration, scleroderma subtype, smoking status, and autoantibody status. The remaining variables were no longer statistically significant.
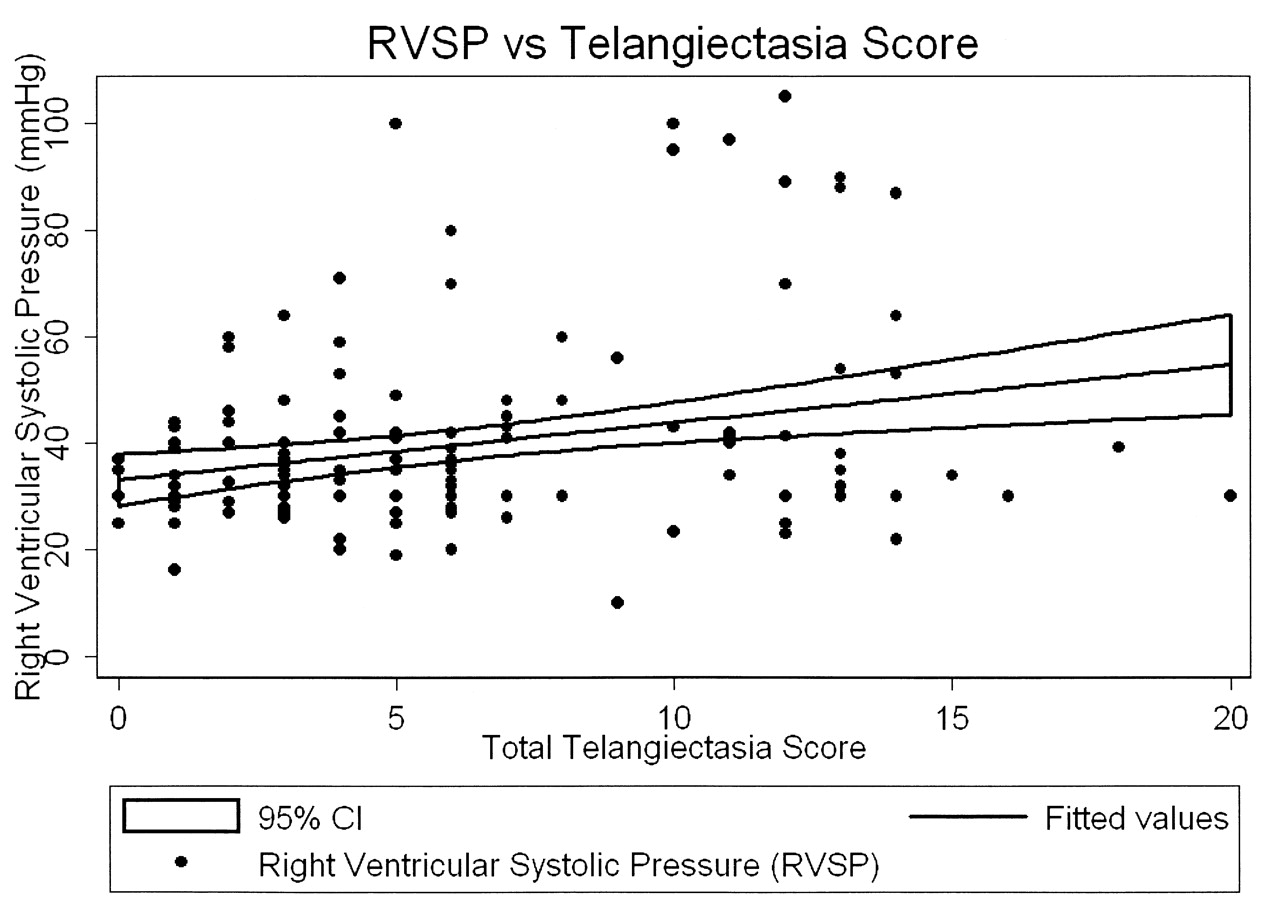
The mean RVSP increased by 10.8 mm Hg for every 10-point increase in telangiectasia score (p = 0.001).
Mean RVSP and DLCO by tertile of telangiectasia score.
Crude and adjusted change in mean RVSP.
Bivariate analysis with logarithmically transformed RVSP yielded similar statistically significant associations for telangiectasia score (p = 0.005), age (p < 0.001), DLCO (p < 0.001), and smoking (p = 0.04), but the association with limited disease subtype was no longer statistically significant (p = 0.14). In multivariable analysis, the mean logarithmically transformed RVSP increased by 0.21 for a 10-point increase in telangiectasia score (p = 0.01), adjusted for age, race, disease duration, subtype, smoking status, and autoantibody status.
Facial telangiectasia score was explored as a possible surrogate marker for the total telangiectasia score. The mean RVSP increased by 4.5 mm Hg for each 1-point increase in facial telangiectasia score, and this approached statistical significance (p = 0.07). After adjusting for potential confounders, the mean RVSP increased by 5.2 mm Hg for a 1-unit increase in facial telangiectasia score (p = 0.065).
PAH on RHC and telangiectasia score
Forty-five patients underwent RHC. The MPAP was 32.2 mm Hg and the mean PCWP was 10.9 mm Hg. Twenty-three of the patients who underwent RHC had confirmed PAH, 8 had PVH, and the remainder were normal. The mean total telangiectasia score differed in these 3 groups: 8.6 in the PAH group, 8.1 in the PVH group, and 4.4 among those with a normal RHC (p = 0.01). Although the increase in telangiectasia score comparing PAH patients to those with a normal RHC was statistically significant (p = 0.01), the increased score among patients with PVH compared to those with a normal RHC was not (p = 0.14).
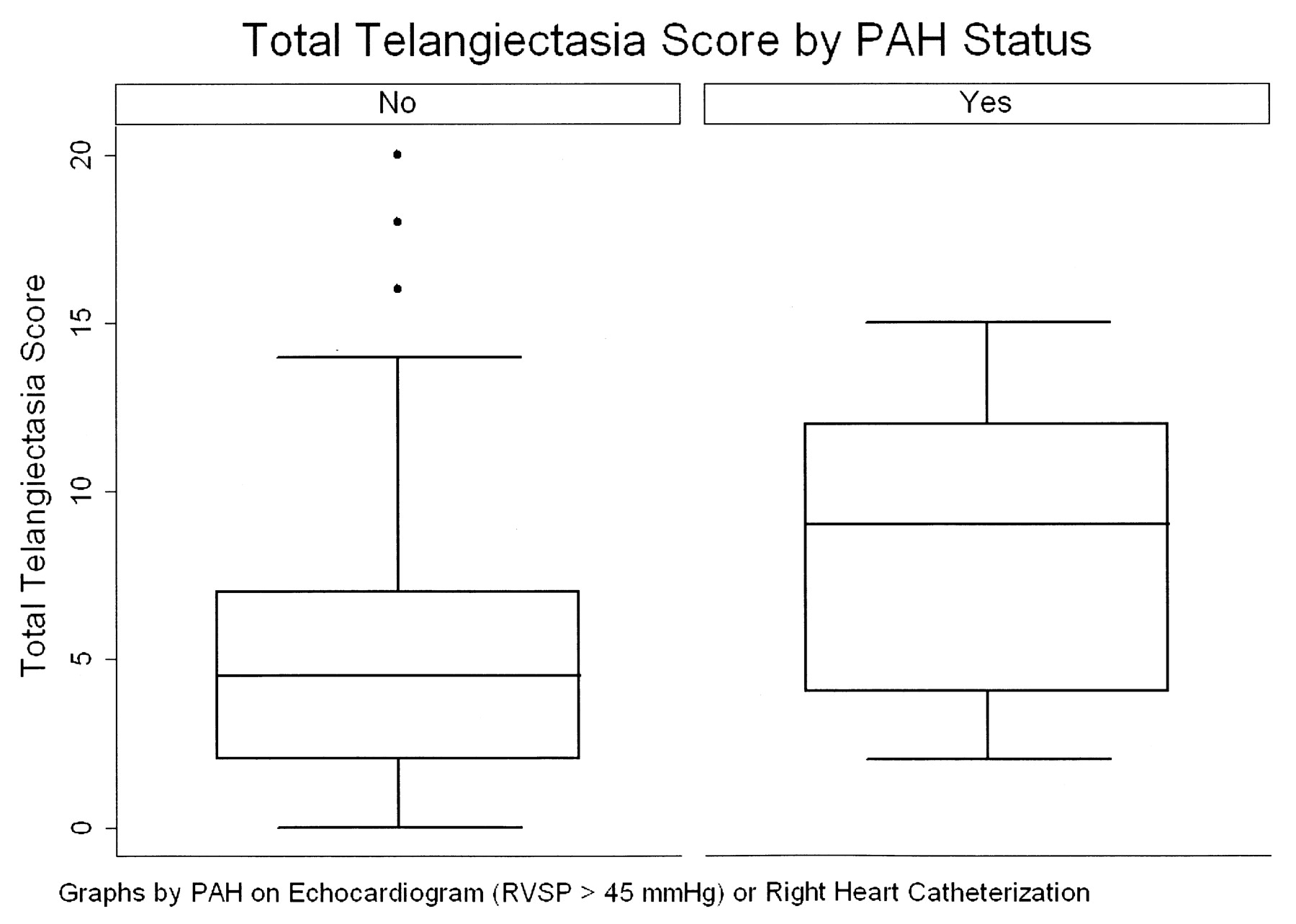
In bivariate analysis, the relative odds of PAH by RHC was 3.9 for patients with a 10-point increase in telangiectasia score (95% CI 1.50–10.0, p = 0.005). In multivariable modeling, the relative odds of PAH were 12.4 for patients with a 10-point increase in telangiectasia score, adjusted for age, race, smoking status, disease duration, scleroderma subtype, autoantibody status, and DLCO (95% CI 1.78–85.9, p = 0.01). Figure 4 shows the distribution of telangiectasia scores among patients without PAH and those with either echocardiographic evidence suggestive of PAH (eRVSP > 45 mm Hg) or RHC-confirmed PAH.

Total telangiectasia score by pulmonary arterial hypertension (PAH) status. PAH is defined as either an estimated right ventricular systolic pressure (RVSP) ≥ 45 mm Hg or right-heart catheterization–confirmed PAH.
Endoglin levels and telangiectasia score
Serum soluble endoglin levels evaluated as part of another study were available in 11 patients. The positive correlation between endoglin levels and telangiectasia score achieved borderline statistical significance in this small sample (r = 0.552, p = 0.078).
Severe Raynaud’s phenomenon and telangiectasia score
Severe Raynaud’s phenomenon (history of digital pitting scars, ulcers, or gangrene) and telangiectasia score were not significantly correlated (r = 0.027, p = 0.46).
DISCUSSION
By quantifying telangiectases in a cross-sectional analysis performed prospectively in a large population of patients with well defined scleroderma, we observed significant associations between evidence of PAH and the number of telangiectases. After adjustment for potential confounders, this association remained robust. Comparing patients with RHC-confirmed PAH to all other patients, the adjusted relative odds of PAH were 12.4 for a 10-point increase in telangiectasia score. The relationship between telangiectasia score and vascular disease appears to be specific to pulmonary vascular disease, rather than a history of severe digital ischemia associated with Raynaud’s phenomenon. In a limited sample of patients, endoglin levels correlated positively with increasing telangiectasia score.
Our results are consistent with a study evaluating telangiectases on the hands10, in which limited scleroderma patients with isolated pulmonary hypertension tended to have multiple telangiectases; however, these authors only evaluated the association of hand telangiectases with pulmonary hypertension in a relatively small sample of patients with limited scleroderma.
Biological evidence for a vascular perturbation driving the pathogenesis of telangiectases has been suggested by increased endothelial proliferation demonstrated on autoradiographic studies of cutaneous telangiectases in patients with scleroderma11. The pathological mechanisms underlying telangiectasia formation have also been studied in hereditary hemorrhagic telangiectasia (HHT), also known as Osler-Weber-Rendu syndrome. This disease is an autosomal-dominant vascular disorder characterized by telangiectases and internal arteriovenous malformations12. The number and distribution of telangiectases are similar in scleroderma and HHT13. In HHT, mutations in portions of the transforming growth factor-ß (TGF-ß) receptor complex have been implicated in causing the disease. Endoglin, a TGF-ß binding protein preferentially expressed on endothelial cells, is responsible for HHT114, and ALK1 (activin receptor-like kinase 1), a type I cell-surface receptor, leads to HHT215. A few studies have investigated the role of endoglin in scleroderma. Telangiectases have been found more frequently in scleroderma patients with elevated soluble endoglin (sENG), and pulmonary artery pressure was found to be positively correlated with elevated endoglin levels16. An association between an ENG gene polymorphism and PAH was suggested in another study17. Elevated levels of circulating sENG were reported in scleroderma patients with cutaneous ulcerations and a low diffusing capacity18. Another study found increased endoglin expression on dermal endothelial cells in scleroderma but no increased serum levels19. These studies may support the theory that telangiectases are an expression of an aberrant systemic vascular process.
It is important to note that given our cross-sectional study design, we were unable to establish a temporal relationship between the presence of telangiectases and the subsequent development of PAH. Further prospective studies are necessary to determine if the number of telangiectases may serve as an early clinical biomarker for the development of severe vascular disease, especially PAH. Taking facial telangiectasia scores may be a possible surrogate measure. It is of interest that the number of telangiectases increased with duration of disease and that they were found in both limited and diffuse skin disease. This pattern suggests that the evolution of new telangiectasia may be a manifestation of ongoing vascular perturbation in scleroderma, and the presence of increased numbers may predict later more severe vascular disease.
Our study showed a strong association between increased numbers of telangiectases and elevated estimated RVSP and RHC-confirmed PAH. Cutaneous telangiectases may be a manifestation of the vasculopathy of scleroderma that could serve as a clinical biomarker for pulmonary vascular disease.
Acknowledgments
The authors thank Adrianne Woods and Cynthia Anderson for assistance in data management and Pamela Hill for assistance in manuscript preparation. We also acknowledge Mary Jo Mulligan Kehoe, PhD, and Michael Simons, MD, for their work on the soluble endoglin levels.
Footnotes
-
Supported in part by the Scleroderma Research Foundation, the American College of Rheumatology Research and Education Foundation’s Clinical Investigator Fellowship Award (Dr. Shah), and by the following NIH grants: 1K23AR052742 (Dr. Hummers) and 1P50 HL084946-01 (Drs. Wigley and Hummers).
- Accepted for publication July 15, 2009.








{kind=link}
{kind=link}
{kind=link}
{kind=link}