


Abstract
L-826,141 [4-{2-(3,4-bis-difluromethoxyphenyl)-2-{4-(1,1,1, 3,3,3-hexafluoro-2-hydroxypropan-2-yl)-phenyl]-ethyl}-3-methylpyridine-1-oxide] is a selective and potent inhibitor of phosphodiesterase 4 (PDE4) with an IC50 value of 0.26 to 2.4 nM for inhibition of the catalytic activity of PDE4A, B, C, and D. The cAMP elevation that can be maintained by PDE4 inhibitors attenuates the signaling cascades that lead to the production of certain cytokines. In cellular-based assays, L-826,141 transcriptionally down-regulates production of tumor necrosis factor (TNF)-α in peripheral blood mononuclear cell and whole blood assays with IC50 values of 31 and 310 nM, respectively. Profiling the effect of this compound on various cytokines in the signaling cascade attenuated by cAMP elevation demonstrates that L-826,141 is also a potent inhibitor of interleukin (IL)-12, granulocyte macrophage-colony stimulating factor, and interferon (IFN)γ (IC50 values of 0.3-0.9 μM) as well as TNF-α formation. We have also shown that the PDE4 inhibitors rolipram and L-826,141 are potent inhibitors of CD3-plus CD28-stimulated IL-2 production in naive human T cells. To address the effect of PDE4 inhibitors on cytokine release from T helper (Th)1 and Th2 effector cells, we used a well characterized model in which T cells are derived from ovalbumin (323-339)-specific T cell receptor transgenic mice. L-826,141 inhibits Th0-mediated IL-2 production with an IC35 value of 25 nM and Th1-mediated IFNγ production with an IC30 value of 46 nM. In contrast, L-826,141 had no significant inhibitory effect (IC30 value > 2.5 μM) on Th2 cell-mediated IL-4 nor IL-13 production. Together, these data demonstrate that specific inhibition of PDE4 preferentially blocks the production of Th1 versus Th2 effector cytokines in vitro.
The second messenger cAMP and the ensuing signaling cascades have been investigated for more than 50 years (Robison et al., 1968). cAMP is generated enzymatically by the action of adenylate cyclase, and this enzyme is activated after the interaction of various ligands with G protein-coupled receptors (Krupinski et al., 1989; Iyengar, 1993; Cooper et al., 1995; Taussig and Gilman, 1995; Sunahara et al., 1996). One example of the G protein-coupled receptor that activates Gαs to stimulate adenylate cyclase activation is the β2 adrenergic receptor (Perry et al., 2002). β2 Adrenergic agonists have been used in clinical practice for the treatment of bronchial asthma for more than 25 years. It has been demonstrated that cAMP signaling may have various downstream effects due to compartmentalization of cAMP pools (Houslay and Milligan, 1997). Because degradation of cAMP occurs through the action of phosphodiesterases (PDEs), several attempts have been made to clinically develop inhibitors of PDEs. The identification and molecular cloning of several gene families of PDEs within the last decade has led to the major focus on novel treatments for asthma and COPD by targeting the PDE4 gene family (Huang et al., 2001). The PDE4 gene family consists of four major genes, A, B, C, and D, with many splice variants (Houslay and Adams, 2003). PDE4 is expressed in the majority of inflammatory cells and its inhibition attenuates the production of many inflammatory cytokines and mediators. Most of the early prototype PDE4-selective inhibitors such as rolipram suffered from various untoward effects such as emesis (Robichaud et al., 2001). All of the PDE4 inhibitors reported to date are either nonselective for any of the four PDE4 genes, A, B, C, or D, or are slightly more potent at inhibiting PDE4D over the other members of this family (Conti et al., 2003). The recent data generated from the PDE4 null mice demonstrate that the bronchodilatory role attributed to PDE4 ablation is mainly through PDE4D, whereas part of the anti-inflammatory role is through PDE4B (Hansen et al., 2000; Jin and Conti, 2002). Specifically, mononuclear cells have been shown to have a reduced capacity to produce TNF-α in the PDE4B (-/-) mice, and PDE4D (-/-) mice are not responsive to muscarinic cholinergic stimulation.
Presently, the most clinically advanced PDE4 inhibitors are roflumilast and cilomilast, which are clinically being developed for the treatment of asthma and/or COPD (Compton et al., 2001; Sturton and Fitzgerald, 2002; Timmer et al., 2002; Profita et al., 2003). A significant effort has been made to delineate the anti-inflammatory properties of PDE4 inhibitors on various cytokines in immune cells. Asthma is often associated with a T helper (Th)2-mediated inflammatory reaction in the airway. The major Th2 cytokines measured in bronchoalveolar lavage (BAL) fluid of patients with asthma are interleukin (IL)-4, IL-5, IL-9, and IL-13, although some Th1 cytokines such as interferon (IFN)γ can also be found in BAL fluid (Prieto et al., 2001). IL-4 and IL-13 have overlapping biological functions due to a shared receptor component and shared signaling pathways (Renauld, 2001). Among other effects, these cytokines also regulate immunoglobulin (Ig)-E class-switching in B cells. IL-4 is required for Th2 differentiation, but ablation of IL-4 alone is not sufficient to block airway hyperreactivity in mouse models of asthma (Hogan et al., 1997). Ablation of IL-13 completely blocks airway hyperreactivity in these models. IL-5 is the key cytokine responsible for the expansion and maintenance of eosinophils (Walter et al., 2001). IL-9 has various effects, including promoting proliferation of bone marrow-derived mast cells. Constitutive expression of IL-9 in the lungs of transgenic mice results in spontaneous airway inflammation characterized by overexpression of other Th2 cytokines (Vink et al., 1999). Granulocyte macrophage-colony stimulating factor (GM-CSF) is a cytokine derived primarily from monocytes/macrophages and stimulates a variety of inflammatory cells, including eosinophils. GM-CSF has also been shown to be elevated in the BAL fluid of patients with asthma after antigen provocation (Broide et al., 1992). COPD, which encompasses emphysema and chronic bronchitis, is characterized by poorly reversible, progressive airway obstruction, mucosal and submucosal inflammatory cell infiltration, edema, fibrosis, mucous plugs, and smooth muscle hypertrophy. BAL fluid and sputum from COPD patients contain increased numbers of neutrophils (in contrast to asthma where eosinophils predominate) and inflammatory mediators such as leukotriene B4 (a major neutrophil chemoattractant and activator), TNF-α, and IL-8 (Barnes, 2003). PDE4 inhibitors have been shown to inhibit many of these cytokines involved in both asthma and COPD. One of the major issues has been which in vitro or ex vivo readout of cytokine inhibition is most relevant for the clinical correlate of efficacy. In this study, we have used human peripheral blood and purified human T cells along with murine polarized T cells to delineate a profile of inhibition of PDE4 compounds.
Materials and Methods
PDE4 Activity Assay. The hydrolysis of cAMP by the purified human recombinant glutathione-PDE4A248 and its PDE4B, PDE4C, and PDE4D equivalents were monitored by following the hydrolysis of [3H]cAMP to [3H]AMP at room temperature in a buffer containing 0.1 μM [3H]cAMP (specific activity 1 μCi/ml), 10 mM MgCl2, and 50 mM HEPES (pH 7.2) as described previously (Laliberte et al., 2000). IC50 values were estimated using a four-parameter nonlinear regression analysis of a 11-point dose-response curve performed in duplicate. The PDE4 enzymes used are the recombinant QT versions of PDE4A, B, C, and D, which encode the catalytic domain of PDE4 and have been shown to be in a fully activated conformation (Liu et al., 2001). The PDE4 constructs termed QT versions were generated by creating glutathione fusion constructs in frame with the Gln-Thr (QT) located within UCR2 of the PDE4A4, PDE4B2, PDE4C2, and PDE4D3 sequences. These constructs were expressed in Sf9 cells and purified to homogeneity as described previously (Laliberte et al., 2000). The purified enzymes were electrophoresed and confirmed to be the predicted molecular mass based on this fusion construct. The IC50 values obtained for the truncated QT versions were also compared with purified full-length enzyme and found to be within 2- to 3-fold that obtained with the corresponding full-length versions of the PDE4A4, B2, C2, and D3 enzymes.
PDE Selectivity Assays. L-826,141 was counterscreened versus PDE1-11 for inhibition of any other phosphodiesterase isoform. PDE1, PDE3, PDE7, PDE8, PDE10, and PDE11 were all assayed similar to the PDE4 enzymes at substrate concentrations of 0.1 μM cAMP. All enzymes were obtained from human recombinant sources except PDE1, which was from dog heart, PDE6 from bovine eye, and PDE2 from human platelets. PDE2 from human platelets was assayed by MDS Pharma Services (Bothell, WA) at 1 μM cAMP. The PDE5, PDE6, and PDE9 enzymes were all assayed at 0.1 μM cGMP.
Cytokine Production in Human Whole Blood. Heparinized whole blood from healthy volunteers was collected and treated as described previously (Muise et al., 2002). Briefly, 500-μl aliquots of blood were preincubated with either 2 μl of DMSO or PDE4 inhibitors at 37°C for 15 min. This was followed by incubation with 10 μl of 0.1% bovine serum albumin, LPS (from Escherichia coli serotype 0111:B4, 1 μg/ml final concentration; Sigma-Aldrich, St. Louis, MO) or concanavalin A (Con A) (50 μg/ml final concentration; Sigma-Aldrich) for the indicated times at 37°C. At the end of the incubation, samples were centrifuged at 1500g at 4°C for 10 min to collect plasma or erythrocytes were lysed using EL buffer (QIAGEN, Chatsworth, CA), and white cells pellets were resuspended in TRIzol reagent (Invitrogen, Carlsbad, CA) for total RNA isolation. Plasma levels of the following cytokines were quantified by ELISA: IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-13, IFNγ, GM-CSF, transforming growth factor-β (BioSource International, Camarillo, CA) and TNF-α (Cistron Biotechnology, Pine Brook, NJ) according to the manufacturers' instructions. Total RNA was first isolated using TRIzol reagent and then cleaned up using the RNeasy mini kit combined to a DNase treatment (QIAGEN) as described in the manufacturer's instructions. Total RNA was reverse transcribed and cytokine mRNA levels were quantified by real-time quantitative PCR as described below. Reverse Transcription and Real-Time Quantitative PCR. Reverse transcription of RNA (50 ng) was performed using Taqman transcription reagents (Applied Biosystems, Foster City, CA). The reaction was performed in the presence of 1× Taqman reverse transcription buffer, 5.5 mM magnesium chloride, 500 μM each dNTP, 2.5 μM random hexamers, 0.4 U/μl RNase inhibitor, and 1.25 U/μl Multiscribe reverse transcriptase. Every reaction set included RNA samples incubated in absence of Multiscribe reverse transcriptase to serve as controls for genomic DNA contamination. The cycling parameters consisted of a primer incubation of 10 min at 25°C, and a reverse transcription of 30 min at 48°C followed by inactivation at 95°C for 5 min. Real-time quantitative PCR reactions were performed on an ABI Prism 7700 sequence detection system (Applied Biosystems) with predeveloped primers and probe sets for all the cytokines mentioned above as targets and 18S rRNA as an endogenous reference purchased from Applied Biosystems. These predeveloped probe/primers sets have been optimized to have similar amplification efficiencies. Each PCR reaction was performed in a total volume of 50 μl, containing 5 μl of cDNA, 25 μl of Taqman Universal PCR Master Mix (Applied Biosystems), 2.5 μl of the commercial probe/primers set, and nuclease-free water (Ambion, Austin, TX) to complete. The cycling parameters consisted of 2 min of uracil removal incubation at 50°C, 10 min of polymerase activation at 95°C, and 50 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min. Every reaction set included a reaction where water was used instead of cDNA to serve as a control for DNA contamination and probe degradation.
Quantitation of Gene Expression. Because predeveloped probe/primers sets have been optimized to have similar amplification efficiencies, the relative quantification of the real-time PCR results was performed using the comparative 2-ΔΔCt method as described previously (Livak and Schmittgen, 2001). Briefly, PCR reactions were performed in parallel for all samples. Measured fluorescent signal intensities were plotted against the number of PCR cycles on a semilogarithmic scale. The cycle number where the PCR amplification was in its exponential phase was referred as the threshold cycle (Ct). To normalize for input amounts of RNA, PCR reactions for the target genes and for the 18S rRNA endogenous reference were performed in separate tubes for each sample and the 18S rRNA Ct values were subtracted from the target Ct values to obtain normalized ΔCt values. The calibrator was defined arbitrarily as the samples preincubated with DMSO and stimulated with 0.1% bovine serum albumin and the normalized ΔCt values of the calibrator was subtracted from the normalized ΔCt values of all the samples to obtain ΔΔCt values. The relative expression of each sample to the calibrator was obtained by calculating 2-ΔΔCt.
CD3/CD28-Dependent IL-2 Secretion in Isolated Human T Cells. Whole blood from healthy volunteers was collected into Vacutainers CPT cell preparation tubes (BD Biosciences, Franklin Lakes, NJ) for the isolation of mononuclear cells. The tubes were centrifuged at 1800g at room temperature for 20 min, and the whitish layer containing mononuclear cells and platelets was collected. Cells were washed with phosphate-buffered saline, and residual erythrocytes were lysed using a buffer containing 0.14 M NH4Cl and 17 mM Tris-HCl, pH 7.5, at room temperature for 7 min. Cells were then centrifuged at 200g for 10 min to remove platelets, and B cells were depleted using anti-CD19 microbeads (Miltenyi Biotec, Auburn, CA) on the AutoMACS instrument (Miltenyi Biotec, Auburn, CA). At the end, cells were resuspended at 2 × 106 cells/ml in Cyto SF-4 medium (Kemp Biotechnologies, Frederick, MD) and dispensed to the wells of a 96-well plate precoated with 500 ng of anti-CD3 (2.5 μg/ml final; BD Biosciences PharMingen, San Diego, CA). Then, cells were pre-incubated with either 1 μl of DMSO or PDE4 inhibitor at 37°C for 15 min. This was followed by the addition of 2 μg/ml anti-CD28 (Bio-Source International) and incubation at 37°C for 18 h. The cell culture supernatant was collected and IL-2 was measured by ELISA (BioSource International).
Murine T Cell Activation and Polarization. Ovalbumin (OVA)-specific TCR-transgenic mice (DO11.10 mice) (Murphy et al., 1990) and BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Splenocytes were prepared from 2- to 6-month-old DO11.10 or BALB/c mice in Leibovitz's L-15 culture medium supplemented with 5% heat-inactivated FCS, 10 mM HEPES, and 50 μg/ml gentamicin. Red blood cells were eliminated by resuspending the cells in 2 ml (per spleen) of ACK lysing buffer (formula 79-0422DG; Invitrogen) for 2 min. The cell suspension was then diluted with a 9× volume of high phosphate-buffered saline with 3% FCS to stop the lysing reaction and remove the dead cells. The cell suspension was centrifuged, and the cell pellet was resuspended in L-15 medium and filtered through a Nitex nylon mesh. The purified splenocytes were then washed two more times in L-15 medium before being resuspended in RPMI 1640 culture medium (Invitrogen) supplemented with 10% heat-inactivated FCS, 1 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 50 μM 2-mercaptoethanol, and 50 μg/ml gentamicin. The cells were cultured in tissue culture flasks for 7 days. The stimuli are as follows: 0.3 μM OVA peptide 323-339, 2 ng/ml m-IL-2, and either Th1 or Th2 cytokines. For generating Th1 cells, 10 ng/ml mIL-12 and 2 μg/ml anti-IL-4 (clone11B11; BD Biosciences PharMingen) were added. For generating Th2 cells, 20 ng/ml mIL-4 and 2 μg/ml anti-IFN-γ (clone R4-6A2) were added. Throughout the polarization, the culture was split to keep the cells in log growth phase and to prevent overcrowding. For a secondary culture, the polarized Th1 or Th2 cells were washed three to four times with L-15 or RPMI 1640 culture medium. The T cells (2 × 105/well) were then cultured in 96-well plates with 5 × 105 irradiated antigen-presenting cells (spleen cells from BALB/c wild type mice) and 0.3 μM OVA peptide 323-339. For preparing antigen-presenting cells, the isolated spleen cells were irradiated (3000 rads) and washed one more time with L-15 medium before they were added to the culture. IL-2, IL-4, and IFNγ were measured using an antibody-based capture assay as described previously (Chen et al., 1994) with modifications for IL-4 and IFNγ. For detecting IL-4 in culture supernatants, 60 μl of anti-mouse IL-4 (2 μg/ml, clone 11B11) was used as the capture antibody, and 60 μl of biotinylated anti-mouse IL-4 (1 μg/ml, clone BVD6-24G2) was used as the detecting antibody. For detecting IFNγ, 60 μl of anti-mouse IFNγ (4 μg/ml, clone R4-6A2) was used as the capture antibody and 60 μl of biotinylated anti-mouse IFNγ (1 μg/ml, clone XMG1.2) was used as the detecting antibody. The data are shown as the mean of triplicate 96-well cultures and the standard error were less than 20% in all cases.
Results
L-826,141 Is a Potent and Selective Inhibitor of PDE4. These studies focused on the molecular characterization of the selective PDE4 inhibitor L-826,141 whose structure is shown in Fig. 1. Comparisons with several other PDE4 inhibitors, including (R)-rolipram (Semmler et al., 1993), roflumilast (Hatzelmann and Schudt, 2001), and its N-oxide metabolite were also performed. The structure of L-826,141 is presented in Fig. 1, and data on the comparison of inhibitors with PDE4A, B, C, and D are depicted in Table 1. L-826,141 is a potent inhibitor of PDE4 (IC50 = 0.26-2.4 nM) and is slightly more potent (at least 3-fold) at inhibiting PDE4B and PDE4D compared with PDE4A and PDE4C (Table 1). This is in contrast to (R)-rolipram, which is equipotent for inhibition of PDE4A, B, and D and is 7.5-fold less potent for PDE4C. Roflumilast and its active metabolite (roflumilast N-oxide) is also slightly more potent for PDE4A, B, and D versus PDE4C (3-10-fold). L-826,141 is at least 450-fold selective for PDE4 because potency versus PDE1-11 were tested and the only appreciable inhibition achieved below 10 μM was with PDE3A and PDE3B (IC50 = 2.1 and 1.1 μM, respectively) and PDE8A (IC50 = 4 μM) (data not shown).

Structure of L-826,141.
Intrinsic potency of PDE4 inhibitors in enzymatic and cell-based assays
The PDE4 enzymes are constructs that express the catalytic domain of the PDE4 enzymes (QT) and have been previously described (Liu et al., 2001, see Materials and Methods). All enzyme assays were performed with 0.1 μM CAMP as described under Materials and Methods. The IC50 values for inhibition of the four PDE4 isoforms were performed at [S] < < Km and are therefore approximate Ki values. Cell-based assays were also utilized in which LPS-stimulated production of TNF-α was measured in the presence or absence of inhibitor.
L-826,141 was also tested versus various substrate concentrations of cAMP and demonstrates clear competitive inhibition for PDE4 with the Km app demonstrating linearity with the inhibitor concentration (Fig. 2). The Km based on these data are 2.6 μM for cAMP, which is consistent with a prior published report (Laliberte et al., 2000), and the Ki is 0.5 nM for L-826,141. It has also been previously demonstrated that the binding of some inhibitors to PDE4 is dependent on Mg2+, and the low-affinity rolipram binding site corresponds to a rolipram/PDE4 complex lacking the Mg2+ ion (Liu et al., 2001). Based on these latter data, we have performed 3H-rolipram binding assays with PDE4 in the absence and presence of Mg2+, and L-826,141 had an IC50 value of 2.2 nM for inhibition of PDE4A with Mg2+ and in the absence of Mg2+ the IC50 value was >1 μM. These data suggest that L-826,141 is a more potent inhibitor of the PDE4 holoenzyme form compared with the apoenzyme form. As a control for this last experiment, rolipram was also tested, and the IC50 value shifted from 5.6 to 260 nM for inhibition of PDE4 in the presence or absence of Mg2+, respectively, similar to previous results (Liu et al., 2001).
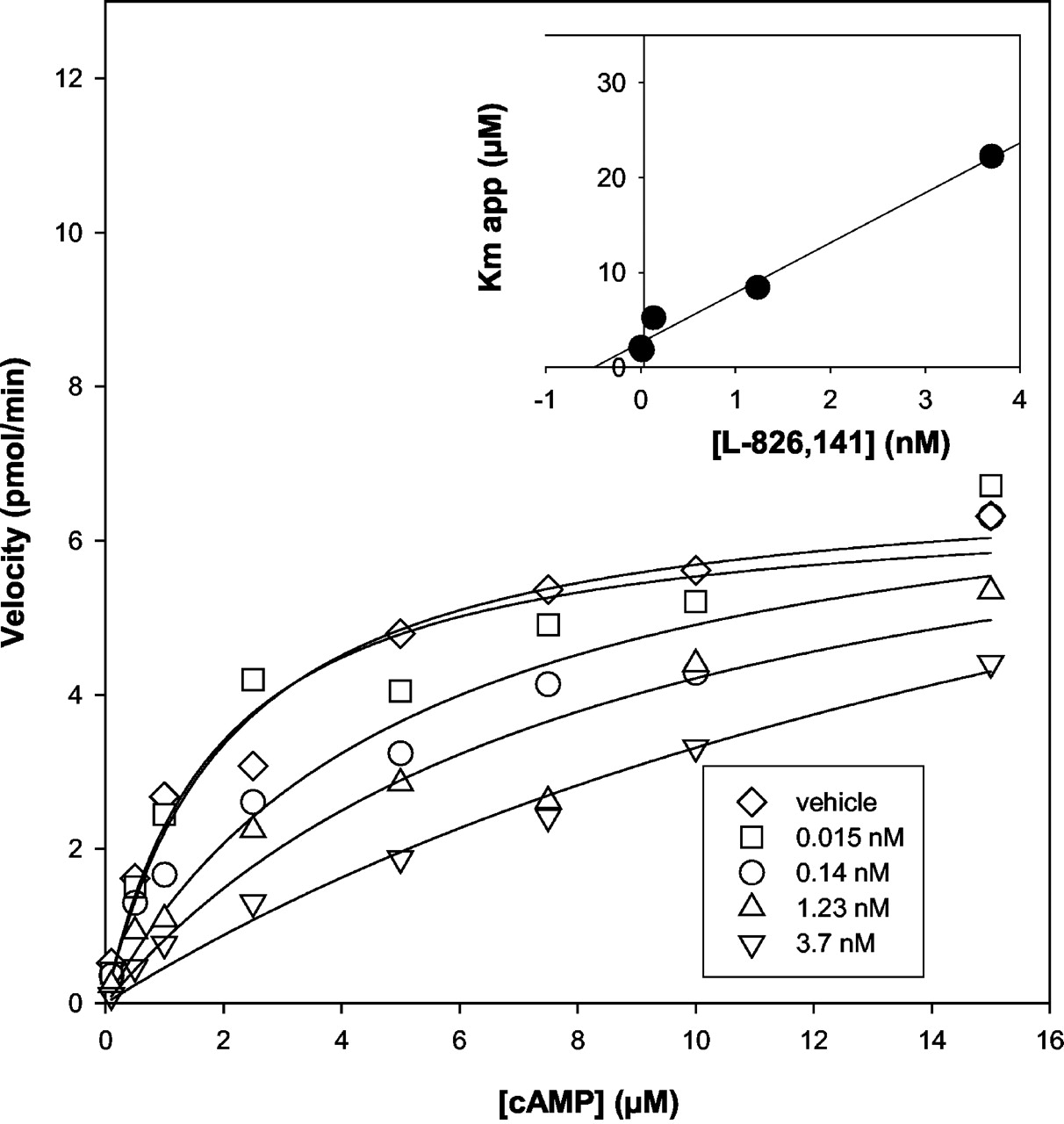
Inhibition of PDE4A activity at various cAMP concentrations. The PDE4A QT enzyme was incubated with either DMSO vehicle or several concentrations of L-826,141 at cAMP concentrations between 0.1 and 15 μM. The specific activity of PDE4A is plotted versus the various cAMP substrate concentrations, and the inhibitor concentrations are depicted in the legend.
Because PDE4 is widely expressed in inflammatory cells, we have also profiled these PDE4-selective inhibitors in human cellular assays using mononuclear cells and peripheral blood. cAMP elevation has been demonstrated to down-regulate production of various cytokines through activation of PKA and indirect mechanisms, including transcriptional down-regulation of the nuclear factor-κB pathway, which can lead to inhibition of the mitogen-activated protein kinase cascade (Simonds, 1999). TNF-α is one of the cytokines downstream of cAMP elevation and mitogen-activated protein kinase activation, which has been used as a surrogate for PDE4 inhibition in the clinic (Timmer et al., 2002). The rank order of potency in the mononuclear cell assays stimulated with LPS to produce TNF-α is roflumilast > roflumilast N-oxide > L-826,141 > rolipram (Table 1). This rank order of potency is maintained in a peripheral blood assay performed in the presence of complete serum. Also, comparing the inhibition of TNF-α production in peripheral blood, the potency of L-826,141 (IC50 = 0.3 μM) is within 2-fold that of roflumilast and its N-oxide and 7-fold more potent than (R)-rolipram (Table 1). These data demonstrate that in cellular-based assays, L-826,141 and (R)-rolipram are least shifted in the presence of high levels of serum protein. Using the mononuclear cell assay, we have also tested the effect of the PKA inhibitor H89 on PDE4 inhibition. The IC50 value for L-826,141 is shifted from 31 nM to 12.4 μM in the presence of 10 μM H89 and demonstrates that TNF-α inhibition is dependent on PKA activation. To confirm that this inhibition is due to cAMP elevation in whole blood, we have used the cell-permeable analog of cAMP, dibutyryl cAMP, and this compound inhibits TNF-α formation in whole blood with an IC50 of 6.5 ± 2.7 μM.
Analysis of Cytokine Production in Peripheral Blood. Human peripheral blood serves as an easily accessible source of myeloid and lymphoid cells for analysis of cytokine production in a clinical setting. We have used peripheral blood under a Toll-like receptor 4 stimulation paradigm with LPS and a nonspecific T cell stimulation with Con A. This study focused on transcriptional down-regulation of key cytokines involved in inflammation and the attenuation achieved by L-826,141. The cytokines analyzed were IL-1β, IL-4, Il-5, IL-6, IL-8, IL-10, IL-12, IL-13, IFNγ, GM-CSF, and TNF-α. The formation of these cytokines at various time points with either an LPS or Con A stimulus was determined by RNA analysis using real-time quantitative PCR. The cytokines that were attenuated by L-826,141 are depicted in Fig. 3 with respect to production over time. Because the 4-h time point resulted in maximal stimulation of cytokine production under the conditions tested, this time point was used for subsequent measurements of cytokine inhibition. IL-1β, IL-5, IL-6, IL-8, and IL-10 were not significantly attenuated by L-826,141 (data not shown). The IC50 values for inhibition of TNF-α, IL-12, GM-CSF, and IFNγ by L-826,141 range from 0.28 to 0.88 μM (Fig. 4; Table 2). Using these conditions, a slight inhibition of both IL-4 and IL-13 was also obtained (Fig. 4), but the maximal mean inhibition achieved was less than 50% at a concentration of 27 μM. IL-2 inhibition was also obtained but was more variable than that observed for the other cytokine measurements. Therefore, IL-2 production and inhibition in isolated human naive T cells was performed after T cell receptor stimulation.

Time course of cytokine mRNA formation in human whole blood. Heparinized whole blood was incubated with LPS (A) or Con A (B) and analyzed over a 24-h period. At the indicated time, total RNA was isolated, reverse transcribed, and analyzed by real-time quantitative PCR analysis. The levels of expression of TNF-α, IL-12p40, GM-CSF, IFNγ, and IL-13 are expressed in mRNA quantity relative to that of the whole blood at time 0, after normalization to 18S rRNA. Data are presented as the mean ± S.E.M. of n = 4 experiments performed in duplicate unless otherwise indicated (†, n = 3).

Inhibition of cytokine mRNA formation by L-826,141 in human whole blood. Heparinized whole blood was preincubated with L-826,141 or vehicle for 15 min. After the preincubation, whole blood was challenged with either LPS (A) or Con A (B) for 4 h. At the end of the incubation, total RNA was isolated, reverse transcribed, and analyzed by real-time quantitative PCR analysis. Relative TNF-α, IL-12p40, GM-CSF, IFNγ, and IL-13 mRNA levels were determined and reported as a percentage of inhibition of the control reaction. Data are presented as the mean ± S.E.M. of n = 3 experiments performed in duplicate unless otherwise indicated (†, n = 2).
Inhibition of peripheral blood cytokines by the PDE4 inhibitor L-826,141
Heparanized whole blood was preincubated with L-826,141 for 15 min prior to LPS or ConA challenge. Four hours poststimulation, the cells were harvested, and RNA was extracted and analyzed by quantitative PCR. Individual data points are presented in Fig. 3.
Inhibition of IL-2 Production in CD3/CD28-Stimulated Human T Cells. IL-2 is the major cytokine involved in proliferation of T cells, B cells, and natural killer cells upon activation of a naive T cell in the presence of a professional antigen-presenting cell. It has been previously demonstrated that IL-2 production can be attenuated by the elevation of cAMP through a PKA-dependent mechanism (Li et al., 1999). To determine the role of PDE4 in IL-2 production in this paradigm, we have isolated naive human T cells from peripheral blood that are CD27+ and CD69- to 87% homogeneity. These cells were challenged with either CD3 alone, CD28 alone, or CD3 plus CD28 combined (Fig. 5) at various time points. Only the CD3 plus CD28 costimulation provided a significant production of IL-2 above the background with a maximal IL-2 production of 3.5 ng/ml. The maximal detectable IL-2 formation was between 8 and 12 h post-CD3/CD28 stimulation. This population of human T cells was then used to determine the PDE4 component responsible for regulating IL-2 production. The early generation PDE4 inhibitor, (R)rolipram, or L-826,141 was preincubated with human T cells before CD3 plus CD28 costimulation and production of IL-2 was measured at 8 or 18 h postantibody challenge. (R)-Rolipram was a potent inhibitor of IL-2 production with an IC50 of 0.4 μM (Fig. 5). The IC50 correlates well with a similar but separate experiment performed in mononuclear cells for TNF-α inhibition by rolipram (IC50 = 0.3 μM). Similarly, L-826,141 was found to have an IC50 value of 3 nM for inhibition of T cell-mediated IL-2 reproduction, and this is consistent with its increased potency in mononuclear cell for inhibition of TNF-α formation. Several PDE4 inhibitors have been shown to block the formation of IL-2, including roflumilast (Hatzelmann and Schudt, 2001), and this may suggest that these compounds may truly have an immunomodulatory role in inflammatory disease.

Effects of rolipram on IL-2 formation in human T cells. A, time course of IL-2 secretion in isolated human T cells. T cells were isolated from human whole blood and challenged with anti-CD28, anti-CD3, or both over an 18-h time period. B, inhibition of CD3/CD28-dependent IL-2 secretion by (R)-rolipram in isolated human T cells. Human T cells were preincubated with (R)-rolipram, L-826,141, or vehicle for 15 min. Then cells were incubated with anti-CD3/anti-CD28 for 18 h. At the indicated time, culture supernatants were collected and analyzed for IL-2 production by ELISA. Each data point is an average of duplicates and is reported in B as a percentage of inhibition of the control reaction. The corresponding data for mononuclear cell TNF-α inhibition and IL-2 inhibition is presented in tabular format.
Inhibition of Polarized Murine T Cell Cytokine Production. As shown above, the cytokines attenuated in peripheral blood by PDE4 inhibitors are TNF-α, IL-2, IFNγ, GM-CSF, IL-12, and to a lesser extent IL-13. Because various cell types besides differentiated T cells can produce some of these cytokines, we performed analyses on differentiated and antigen-challenged T cells. Several reports of PDE4 inhibitors have demonstrated attenuation of either Th1- or Th2-mediated cytokine production, depending on the in vitro method used. Because conditions to generate polarized Th1 and Th2 cells have been well established in the murine system, we have decided to perform antigen presentation assays in murine Th0, Th1, and Th2 cells. The results presented from Th0 and polarized Th1, Th2 cells were obtained from ovalbumin-specific TCR transgenic mice. Polarization of Th1 and Th2 cells was performed as described under Materials and Methods, and the antigen used for all three cell types was OVA 323-339 at a concentration of 0.3 μM. Results for L-826,141 in comparison with dexamethasone are presented in Table 3. The compounds were added either after the first antigen challenge for Th0 cell activation or after the second antigen challenge for Th1- and Th2-polarized cells. L-826,141 and dexamethasone inhibited Th0 antigen-provoked IL-2 production. Dexamethasone was the most potent inhibitor with an IC50 value of 12 nM (IC35 = 5 nM), which is consistent with previously reported data (Kavelaars et al., 1995). The inhibition achieved with L-826,141 was maximal at 60 to 70% and with an IC35 value of 25 nM. These data demonstrate that PDE4 inhibitors can attenuate IL-2 production but that the inhibition is partial and less than that seen with dexamethasone. The polarized T cells were analyzed for production of IFNγ, IL-4, and IL-13. To confirm that the T cells were appropriately polarized, Th1 cells were analyzed for IFNγ production and resulted in low or undetectable production of IL-4 and IL-13, demonstrating a polarized population of Th1 cells. Th2 cells were analyzed for formation of IL-4 and IL-13 and produced low or undetectable levels of IFNγ. In antigen-challenged Th1 cells, dexamethasone was a weak inhibitor of IFNγ production with maximal inhibition of 47% at a concentration of 300 nM, consistent with a Th2 profile of inhibition. In the antigen-challenged polarized murine Th1 cells, L-826,141 inhibited IFNγ formation with an IC30 value of 46 nM. Dexamethasone inhibited IL-4 and IL-13 production with IC50 values of 3.5 and 4 nM, respectively, and as seen with IL-2 inhibition the maximal inhibition achieved was between 75 and 85%. In contrast to dexamethasone, L-826,141 was a weak inhibitor of IL-4 and IL-13 formation with L-826,141 achieving only 37% inhibition of IL-4 production at a concentration of 10 μM. Roflumilast and its corresponding N-oxide (data not shown) also did not achieve inhibition of IL-4 greater than 30% in the Th2 cells up to a concentration of 10 μM. Therefore, the selective PDE4 inhibitor L-826,141 can inhibit OVA-challenged Th0 cells to produce IL-2 and has a propensity for inhibition of Th1 versus Th2 cytokine production.
Th0, Th1, and Th2 cells from ovalbumin transgenic mice (DO11)
T cells from ovalbumin-specific T cell receptor transgenic mice (DO11) were isolated and cultured as described under Materials and Methods. Th0 and polarized Th1 and Th2 cells were challenged with 0.3 μM OVA peptide 323-339 in the presence or absence of the inhibitors. The production of the cytokines was determined by immunoassays.
Discussion
PDE4 inhibitors are presently under clinical development for the treatment of asthma and/or COPD. The clinical candidates that are the furthest advanced are cilomilast (Compton et al., 2001) and roflumilast (Timmer et al., 2002). In this study, we have focused primarily on the compound L-826,141 with some comparative in vitro data with (R)-rolipram and roflumilast. These compounds target the PDE4 gene family, which includes PDE4A, B, C, and D along with the various splice variants (Houslay and Adams, 2003) and are competitive inhibitors of the nucleotide binding site for cAMP on PDE4. Recently, the crystal structure of PDE4B and D have delineated the active site of PDE4 and demonstrated that (R)- and (S)-rolipram bind within the cAMP binding domain of PDE4 (Xu et al., 2000; Dym et al., 2002). The earliest precursor to PDE4 inhibitors could be attributed to caffeine and the methylxanthines, but these compounds are very weak PDE4 inhibitors (Horiuchi and Castro, 2000). These compounds, although very ineffective as PDE4 inhibitors, were known to have efficacy in pulmonary diseases. The development of potent and selective PDE4 inhibitors has not yet led to a clinically approved candidate. In this study, we focus on the mechanistic profile of inhibition at the enzyme and inflammatory cytokine level. The compound that has been profiled is L-826,141, which is a structural analog to CDP840, which has previously demonstrated attenuation of the late asthmatic response in humans in an allergen challenge paradigm (Harbinson et al., 1997).
L-826,141 is a potent, selective, and competitive PDE4 inhibitor with IC50 values in the 1 nM range for the four PDE4 isoforms. The inhibitor preferentially inhibits the holoenzyme compared with the apoenzyme. Because PDE4B and D are essential for LPS-stimulated TNF-α formation and smooth muscle hyperreactivity (Hansen et al., 2000; Jin and Conti, 2002), respectively, analysis of intrinsic potency on these enzymes demonstrates that L-826,141 is equipotent to roflumilast N-oxide, which is the relevant metabolite of roflumilast formed in vivo. L-826,141 is also a very potent inhibitor of PDE4A and C, and the role that these latter two enzymes play in inflammation shall only be determined through development of subtype-selective inhibitors or phenotypic analysis of the corresponding null mice.
Because PDE4 is the major enzyme that metabolizes cAMP in inflammatory cells, significant research has focused on the role of PDE4 in Gαs interactions. One suggested role is the interaction with Gαs through the β-arrestin adaptor molecules and desensitization of signaling through the β2 adrenergic receptor (Perry et al., 2002). Because β2 adrenergic receptor activation leads to various signaling cascades and stimulation and/or inhibition of various cytokines, we have used peripheral blood, isolated human T cells and murine polarized T cells to determine the Th1/Th2 balance of inhibition obtained with selective PDE4 inhibitors. These data demonstrate that at nanomolar concentrations L-826,141 is a potent inhibitor of TNF-α, IL-12, GM-CSF, and IFNγ in peripheral blood. Using a CD3/CD28 costimulation of naive T cells, 80% of IL-2 production was inhibited by both selective PDE4 inhibitors, L-826,141 and rolipram. This clearly delineates that the cAMP pool that activates PKA-dependent IL-2 production in isolated T cells is directly linked to the PDE4 gene family. Previously, data had suggested that PDE7 may play an important role in IL-2 generation from T cells (Li et al., 1999). Our data with the PDE4 inhibitors L-826,141 and rolipram and also recent data with PDE7A null mice (Yang et al., 2003) demonstrating that IL-2 levels and T cell proliferation upon CD3/CD28 stimulation are similar in PDE7A(-/-) as in wild-type littermates suggest that PDE7 is not essential for T cell function.
Peripheral blood is skewed toward primarily the production of Th1 cell-associated cytokines; therefore, we also analyzed polarized and antigen-challenged murine T cells to determine the Th1/Th2 balance of inhibition obtained with selective PDE4 inhibitors. The data from polarized T cells confirms the peripheral blood data and demonstrates conclusively that L-826,141 has a greater propensity to inhibit Th1-derived cytokines in vitro and IL-2 production than Th2-derived cytokines. Based on the paradigm that asthma is primarily a Th2-biased immunological disorder and COPD has primarily a Th1 bias, the data generated suggest that compounds such as L-826,141 will have a greater anti-inflammatory impact on COPD- and Th1-based immune disorders. Our data are in contrast to several reports demonstrating that PDE4 inhibitors can attenuate production of IL-4 and IL-5 production in various stimulation paradigms. Ariflo has been reported to inhibit oxazolone-induced IL-4 production in a mouse model of ear inflammation (Griswold et al., 1998). Roflumilast in our hands also demonstrates a lack of inhibition of IL-4 in the in vitro transgenic mouse model of OVA challenge T cells, whereas a literature report has demonstrated that roflumilast inhibits IL-4 and IL-5 production in CD3/CD28 costimulated human T cells (Hatzelmann and Schudt, 2001). Rolipram has also been shown at elevated doses (5-10 μM) to inhibit IL-5 formation in OVA-challenged mouse splenic cells in vitro and IL-4 and IL-5 from BALF in OVA-challenged mice (Foissier et al., 1996). There is also data with rolipram demonstrating that in human T cell antigen presentation assays IFNγ from Th1 cells can be significantly inhibited without altering formation of IL-4 from Th2 cells (Bielokova et al., 2000) consistent with the profile seen with L-826,141. The true utility of PDE4 inhibitors such as L-826,141 can only be determined if a PDE4 inhibitor is clinically approved.
cAMP signaling has been studied for more than five decades, and the complete pathways have not yet been fully elucidated. We have performed analyses on cytokine production to determine the effects of blockade on cAMP metabolism through the action of PDE4 inhibitors. cAMP levels result in various manifestations, including effects on smooth muscle contractility, neurochemical mediator release, chemotaxis, cell adhesion, vasopermeability, mucociliary activity as well as effects on inflammatory cell activity. PDE4 inhibitors clearly have potential as novel new anti-inflammatory agents for several different immune-mediated diseases. The clinical approval of this new class of compounds may hold significant promise in the treatment and control of asthma, COPD, and other immune based diseases.
Footnotes
-
DOI: 10.1124/jpet.103.064691.
-
ABBREVIATIONS: PDE, phosphodiesterase; COPD, chronic obstructive pulmonary disease; TNF, tumor necrosis factor; Th, T helper; BAL, bronchoalveolar lavage; IL, interleukin; IFN, interferon; GM-CSF, granulocyte macrophage-colony stimulating factor; DMSO, dimethyl sulfoxide; LPS, lipopolysaccharide; L-826,141, 4-{2-(3,4-bis-difluromethoxyphenyl)-2-{4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)-phenyl]-ethyl}-3-methylpyridine-1-oxide; Con A, concanavalin A; ELISA, enzyme-linked immunosorbent assay; PCR, polymerase chain reaction; OVA, ovalbumin; FCS, fetal calf serum; PKA, protein kinase A; H89, N-(2-[p-bromocinnamylamino]ethyl)-5-isoquinolinesulfonamide hydrochloride.
- Received December 19, 2003.
- Accepted April 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}