Article Text

Abstract
Objective This 9-month open-label extension of the Circadian Administration of Prednisone in Rheumatoid Arthritis Study (CAPRA 1) investigated the long-term safety and efficacy of prednisone chronotherapy with a novel modified-release (MR) prednisone for up to 12 months.
Methods Of 288 patients with rheumatoid arthritis originally randomised to MR or immediate-release (IR) prednisone, 249 continued with prednisone chronotherapy (2–10 mg/day) in the 9-month open-label extension. Duration of morning stiffness of the joints (MS), disease activity scores (DAS28), American College of Rheumatology (ACR20) responses and plasma levels of interleukin 6 (IL-6) were assessed. Safety was analysed from adverse event reports and laboratory investigations.
Results During the 3-month double-blind phase, patients in the MR group achieved a reduction in MS of 33.1% while no change was observed in the IR group. After 6 months of treatment, MS was reduced in the IR/MR group by 54% and in the MR/MR group by 56%. MS reduction after 12 months was 45% (IR/MR group) and 55% (MR/MR group). Plasma levels of IL-6 declined on MR treatment. DAS28 was reduced from 5.8 to 4.8 (MR/MR group) and 4.9 (IR/MR group), respectively. 37% of the 219 patients who completed the 12-month study achieved improvement according to the ACR20 criteria. Adverse events did not differ from the known profile of low-dose prednisone.
Conclusions Prednisone chronotherapy with the MR tablet was safe and well tolerated and provided a sustained improvement which resulted in a better benefit to risk ratio of low-dose glucocorticoid treatment for at least 12 months.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that can cause joint damage, extra-articular manifestations and disability. Symptoms include fatigue, joint pain and swelling and morning stiffness of the joints (MS), reducing quality of life and the ability to remain gainfully employed.1
Although accepted classification and remission criteria for RA include MS,2 3 this symptom remains neglected in clinical studies. MS was found to be strongly associated with pain and functional incapacity and, to a lesser degree, with swollen and tender joint counts and acute phase responses.1 Severe MS in the early course of RA was identified as having a high impact on patients' early retirement from working life and, therefore, greater attention should be paid to effective treatments of MS.1
The symptoms of RA show pronounced circadian rhythms with the highest severity in the early morning. The redistribution of interstitial fluids while asleep and circadian changes in synovial fluid composition may contribute to stiffness, because oedema of the synovium and the periarticular structures interferes with joint biomechanics.4 Many studies on circadian rhythms in RA have reported the temporal relationship between raised levels of proinflammatory cytokines, insufficient anti-inflammatory protection and RA symptoms.5,–,10 A recent study investigating overnight variations in cytokine and cortisol concentrations in 16 patients with RA reported that nocturnal interleukin 6 (IL-6) levels start to increase approximately 3 h before increases in cortisol and peak at approximately 08:00 h, 40 min before cortisol.11 From all these data, it was concluded that there must be a link between the overnight rise in IL-6 and the circadian variation in RA symptoms.
This conclusion could have important consequences for dose and timing of oral glucocorticoid (GC) treatments. Indeed, it has previously been suggested that GC administration between 06:00 and 08:00 h could be just too late as night-time pathophysiological processes have already initiated inflammation.4 6 Preventing the rise in proinflammatory cytokines should therefore be more efficacious than treating the symptoms. In line with this thought, a modified-release (MR) prednisone tablet was developed to optimise GC therapy of RA in a novel chronotherapy approach. If taken at bedtime, the new tablet releases prednisone 4 h after ingestion—that is, at approximately 02:00 h (Comparative bioavailability study of one new timed-release formulation of prednisone (5 mg tablet) dosed in the evening with or without food and a reference immediate-release formulation (Decortin® 5 mg tablet) dosed in the night without food, after single oral dose administration in healthy male subjects. Clinical Study Report, Merck KGaA, Darmstadt, Germany, 12 December 2003, EMR 62 215-005). The efficacy and safety of this medication was investigated in a controlled clinical study in 288 patients with active RA who were previously treated with GCs, disease-modifying antirheumatic drugs (DMARDs) and non-steroidal anti-inflammatory drugs (NSAIDs).5 During the initial double-blind treatment phase, all patients either continued on immediate-release (IR) prednisone or were switched to prednisone chronotherapy with the novel MR formulation, both at their individual prestudy doses. The primary end point was the relative change in duration of MS after 12 weeks of treatment, which showed significantly greater improvements in the prednisone chronotherapy group. The safety profile did not show differences between the two treatments.5 The additional benefit of the new tablet was considered important for daily clinical practice.12
In this paper we report the results of using low-dose prednisone chronotherapy for up to 12 months.
Methods
Patients
All patients who completed the double-blind study and did not develop any exclusion criteria were eligible to continue treatment with MR prednisone. The study was conducted in compliance with the Declaration of Helsinki at 29 centres in Germany and Poland. The protocol was approved by the responsible administrative bodies and ethics committees.
Study design
The study design, types of treatment and visit schedule are summarised in figure 1A. Before the study all patients had been treated with stable doses of GCs, DMARDs and NSAIDs. During the 3-month double-blind phase, patients received either IR prednisone in the morning and MR prednisone placebo in the evening, or IR prednisone placebo in the morning and MR prednisone in the evening. During the open-label extension all patients received MR prednisone in the evening. The study was performed between August 2004 and January 2007.

Study design and overview of study population. (A) Study phases, treatments and visit schedule; shaded boxes, treatment with MR prednisone open boxes, treatment with IR prednisone. (B) Trial profile. DB, double-blind study phase; IR, immediate-release; MR, modified-release. *Data reported by Buttgereit et al.5
Procedures
The first dose of open-label MR prednisone (Skye Pharma, Lyon, France; distributed by Merck KGaA, Darmstadt, Germany) was taken between 21:30 h and 22:30 h on the last day of the double-blind study. The starting doses for individual patients were equivalent to doses during the double-blind phase. Daily doses were composed of the appropriate number of 1 mg and 5 mg tablets. In contrast to the requirement of stable doses for prednisone and DMARDs during the double-blind phase, dose changes were permitted during the open-label extension. During the double-blind phase, patients completed the appropriate pages in their diaries every morning and every evening before going to bed. During the open phase, completion of the diary pages was required every morning and every evening for at least 7 days immediately before each scheduled study visit. Requested entries included waking-up time, stiffness (yes/no), time of resolution of stiffness and intensity of pain during the day (100 mm visual analogue scale, VAS).
Additional procedures included documentation of adverse events (AEs), blood sampling for erythrocyte sedimentation rate (ESR) and C reactive protein, assessment of the disease activity score (DAS28, including 28 joints checked for swelling and tenderness), patient's assessment of disease activity on a 100 mm VAS (end points 0=not active and 100=extremely active) and physicians' global assessment of disease activity (5-point scale: asymptomatic, mild, moderate, severe, very severe). At each visit investigators recorded all AEs following accepted clinical trial guidelines and definitions. No checklists with predefined AEs were used.
Procedures performed at the end of the study included a physical examination, blood sampling for laboratory investigations and determination of IL-6 plasma concentrations. Local biochemistry laboratories performed ESR measurements, haematology and urinalysis; all other biochemistry analyses were performed by Bio Analytical Research (BARC), Gent, Belgium.
Target variables
Efficacy end points were absolute and relative reductions in MS duration, changes in IL-6 plasma levels, DAS28, pain intensity and American College of Rheumatology (ACR20) improvement. The safety of MR prednisone was analysed from AE reports, clinical biochemistry investigations and physical examinations.
Statistical analysis
MS duration was calculated from the diaries as 7-day means before each visit and defined as the difference between the time of resolution of stiffness and the time of awakening. This was expressed as absolute and relative changes from baseline corresponding to the start of the double-blind phase (xbase). Thus, MS duration was expressed as absolute changes defined as postbaseline value minus baseline value (ie, xvisit – xbase). Relative changes from baseline are given as 100 times the absolute change under treatment divided by the baseline value (ie, 100% × (xvisit − xbase)/xbase).
Data of the open-label phase were evaluated using descriptive statistics.
Although all patients were treated with MR prednisone during the open-label extension, we describe the groups separately as randomised. Accordingly, groups are identified as the IR/MR group with 9 months of treatment and the MR/MR group with 12 months of treatment with MR prednisone.
Results
Of the 288 randomised patients, 251 completed the double-blind phase.5 All qualified for continuation with MR prednisone and 249 patients consented to continue, 219 of whom completed the entire 12-month study. The patient disposition is shown in figure 1B.
Baseline demographics and main disease characteristics did not differ between the 288 patients starting the double-blind phase and the 249 patients continuing MR treatment in the open-label extension (table 1). Of the 288 randomised patients, 272 (94.4%) received concomitant therapy with DMARDs at baseline (MR group: n=133, 92.4%; IR group: n=139, 96.5%). These DMARDs were (in the MR and IR groups, respectively): methotrexate in 74% and 73% of patients; sulfasalazine in 13% in both groups; leflunomide in 11% and 18%; and <10% of patients in both groups had a combination of two DMARDs. Of the 249 patients in the open phase of the study, 238 (95.6%) received DMARDs at baseline. During the open-label extension three more patients (n=241) added a DMARD, increasing the percentage to 96.8%; 76.3% of these 241 patients were receiving methotrexate, 11.9% were receiving sulfasalazine, 14% were receiving leflunomide and <10% were receiving combination therapy. The use of specific DMARDs in the two study phases was therefore similar.
Patient characteristics and disease activity at baseline before start of the double-blind trial phase of the total study population (N=288) and of the group of patients continuing treatment with open-label modified-release prednisone (N=249)
Morning stiffness
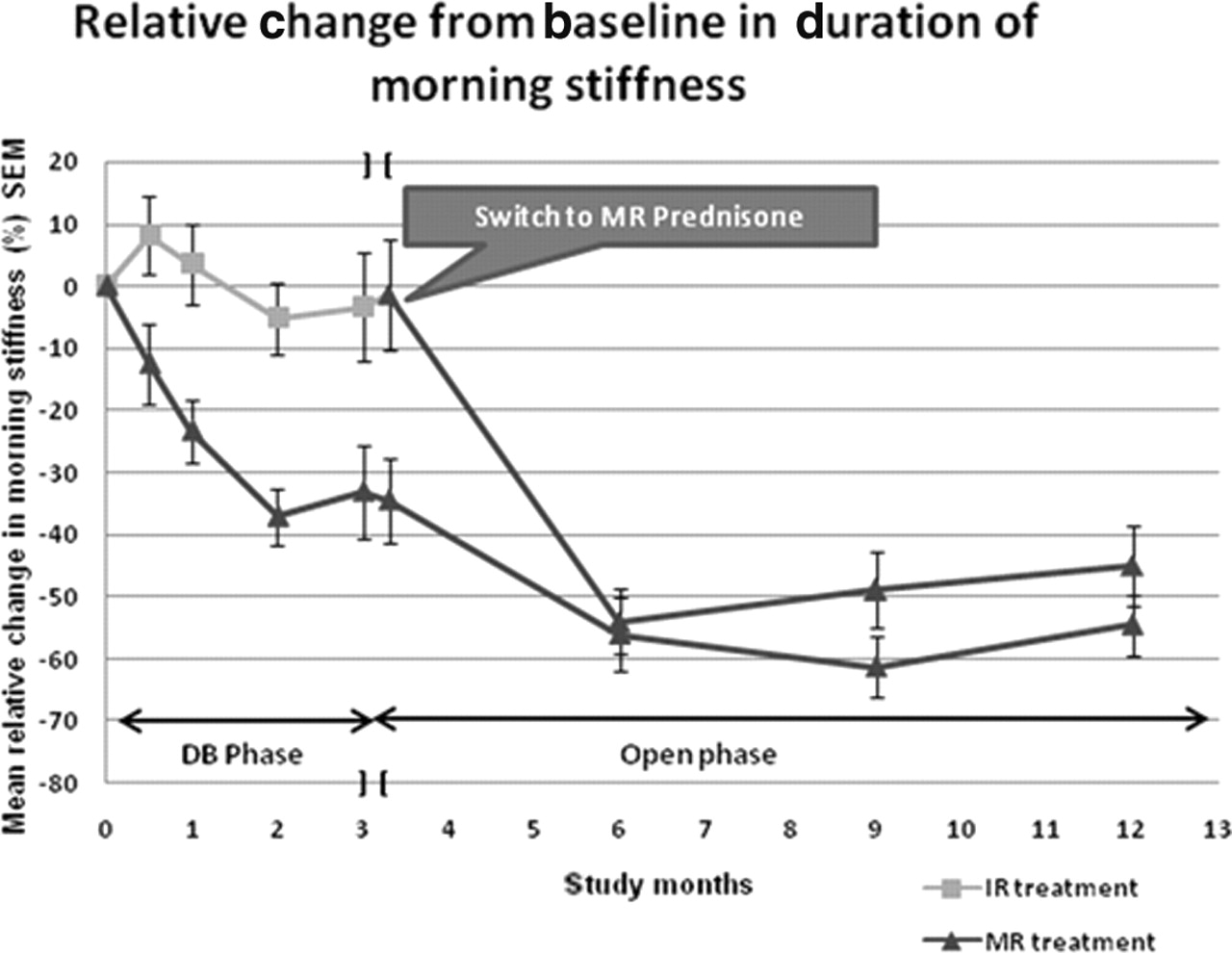
The course of the relative changes in the duration of MS from baseline is shown in figure 2. Owing to the different treatments during the double-blind phase, the MR/MR group started the open phase with MS −34.5% from baseline and the IR/MR patients with MS −1.4%. Two patients who completed the double-blind phase did not enter the extension study, so data at the end of the double-blind phase and the start of the open-label extension are slightly different. When the first assessments were made after 3 months of open-label treatment, MS was reduced from baseline by 56% in MR/MR patients and by 54% in IR/MR patients. The reduction in MS was sustained throughout the entire study period. The reduction in MS corresponded to 49% (IR/MR group) and 61% (MR/MR group) at 9 months and 45% (IR/MR group) and 55% (MR/MR group) at 12 months.
{kind=link}
{kind=link}
Duration of morning stiffness during both study phases. Course of relative changes in duration of morning stiffness for the 288 patients starting the double-blind phase and the 249 starting the open phase after 3 months of double-blind treatments (n=number of available data for baseline (129/125) and end of double-blind phase (111/100), at start of open phase (112/101) and at 6 (95/86), 9 (104/88), 12 months (97/86) for patients in IR and MR groups. DB, double-blind study phase; IR, immediate-release; MR, modified-release.
The mean (SD) observed absolute reduction in MS was 88 (128.3) min in the IR/MR group from a baseline value of 182 (127.4) min. The mean (SD) absolute reduction in MS in the MR/MR group was 83 (83.7) min from a baseline value of 156 (97.3) min. At the end of the 12-month study, 17% of the patients no longer reported MS. The number of patients with duration of MS <1 h increased from 10% at screening (before the start of the double-blind phase on IR prednisone) to 29% at the end of the entire study (on MR prednisone). Accordingly, the number of patients with a longer duration of MS (≥1 h to <3 h and ≥3 h) decreased from 46% to 26% and from 33% to 10% at study completion.
Secondary variables
In the double-blind study the concentrations of IL-6 were almost halved by MR prednisone whereas no changes were observed in the IR group.5 The same reduction by about 50% was seen at the end of the study in the IR/MR patients (from baseline 1110 IU/l to 515 IU/l, median values). Low levels of IL-6 were sustained, but no further reduction was observed in the MR/MR group (median 470 IU/l; table 2).
Summary of secondary efficacy results‡
DAS28 and pain showed clinically relevant improvements with no differences between the treatment groups (table 2). More favourable results were also seen in the patients' and physicians' global assessments of disease activity. Overall, increases in the percentage of patients with mild disease activity (from 9% to 40%) and a decrease in patients with moderate disease activity (from 71% to 52%) were seen from baseline to study end. At the start of the double-blind phase 19% of patients had severe disease activity which was reduced to 7% at 12 months.
Of the 219 patients who completed the entire study, 37% achieved an ACR20 response.
Safety
The median duration of exposure to MR prednisone in the open-label phase was 281 days (range 56–406) and the mean dose was 6.8 mg/day (median 6.4; range 1.9–21.9). The latter takes into account any changes in therapy (eg, temporary interruptions in therapy, dose reductions for reduced disease activity and intermittent dose increases to control flares). Although dose changes were permitted during the open-label extension, no important changes in the ‘stable’ doses were recorded.
Thirty patients (12%) discontinued treatment during the open-label extension (figure 2B). Twelve patients discontinued for AEs. The most frequent reason for discontinuation was RA exacerbation in six patients. This was reported as an AE in both study phases. Other medical reasons for discontinuation were infection (n=5), gastrointestinal tract disorders (n=3) and pregnancy (n=2). The gastrointestinal events were judged by investigators to be possibly related to prednisone, although all three patients were also receiving DMARDs and NSAIDs.
A total of 51 serious AEs were reported by 33 patients. Only two of these events (gastric ulcer perforation and gastrointestinal haemorrhage) were judged by investigators to be possibly related to study medication. However, both patients were also taking DMARDs and NSAIDs. Bone fractures occurred in three patients and tendon rupture in one. These events were not causally attributed to prednisone by the investigators. The other serious AEs reported, including gynaecological problems, cardiovascular diseases, respiratory diseases, joint replacement surgery and synovectomy, were consistent with medical conditions expected for this patient population.
AEs were observed in 51% of the patients. The most frequent events were RA-related symptoms (14.5%), upper respiratory tract infections (2.8%), back pain (2.8%) and weight increase (2.8%). AEs rated as being possibly related to study medication and observed at a frequency of >1.0% in the total study population were upper abdominal pain (n=3, 1.2%), gastritis (n=4, 1.6%) and weight increase (n=6, 2.4%). AEs indicative of aggravated hypothalamic-pituitary-adrenal (HPA) axis suppression were not observed.
Discussion
This study investigated the effects of switching from standard prednisone therapy to prednisone chronotherapy with MR prednisone in patients with active RA over a period of 12 months. In the double-blind phase of the study, MR prednisone was found to produce a significantly greater reduction in the duration of MS than IR prednisone and a significant reduction in IL-6 levels.5 The reduction in MS duration and IL-6 levels established during the double-blind phase in the MR group (MR/MR) was found to be sustained during treatment with MR prednisone for up to 12 months. Similar improvements were also evident in patients switched to MR prednisone in the open-label extension. Clinically relevant improvements in disease activity (DAS28) and pain intensity (VAS) were also observed over the 12 months. The results of our study thus demonstrate that the efficacy of GC therapy can be improved by administration as chronotherapy—that is, targeting drug release to the circadian rhythm of the underlying inflammation and resulting symptoms. This is an important observation given the widespread clinical use of GCs in the treatment of RA and other rheumatic diseases.
It is encouraging to see that 37% of the patients reached an ACR20 improvement at the end of the study despite long-term GC pretreatment and longstanding disease. This is of special importance because, with a disease history of ≥10 years as seen in our patients, pain and other symptoms are not only caused by inflammation but also, at least in part, by irreversible damage to the synovium, cartilage and bone. Nevertheless, MR prednisone with its convenient night-time administration regimen successfully reduces the inflammatory component in a unique way.
The results of this study also further confirm the low level of clinically relevant AEs associated with low-dose GC therapy and are in agreement with published reviews.13 14 MR prednisone was found to have a similar safety profile to IR prednisone given at the same dosages. In particular, no signs or symptoms were noted that might indicate aggravation of suppression of the HPA axis. Indeed, a study of HPA axis function in a subgroup of patients from this study reported elsewhere15 found that: (1) cortisol levels immediately before and after stimulation were comparable between treatments and (2) there was no evidence of greater suppression of the HPA axis during treatment with MR prednisone for up to 12 months. Given the superior efficacy reported from the double-blind study for MR prednisone over IR prednisone and the similar safety profile, MR prednisone can thus be expected to have an improved benefit to risk ratio over IR prednisone.
A potential limitation of the study is that it involved two differently designed phases: an initial blinded active controlled phase followed by an uncontrolled open-label extension. Therefore, only descriptive statistical methods could be employed to analyse data from the open-label extension. However, the IR prednisone group showed comparable effects when switched to MR prednisone chronotherapy, thereby confirming the robustness of the observed treatment effects.
In conclusion, the results of the completed CAPRA1 study reported here suggest that low-dose MR prednisone chronotherapy offers significant benefits over IR prednisone for the treatment of RA which are maintained for up to 12 months. Further studies are warranted to investigate the benefit of low-dose MR prednisone chronotherapy in both GC-naïve patients with early RA and those with other inflammatory conditions. Indeed, chronotherapy may well offer significant advantages over standard therapies in diseases such as polymyalgia rheumatica16 17 and asthma.
Acknowledgments
Biostatistician Ursula Tilp, Premier Research Group, performed the statistical analyses. Christine Knauer, NitecPharma GmbH, created all the figures and tables and undertook careful proofreading of the text.
References
Footnotes
Investigators Germany F Buttgereit, co-ordinating investigator for entire study, Germany (Charité University Medicine, Berlin), R Alten, co-ordinating investigator for CRH test substudy (Schlosspark-Klinik, Berlin), E Gromnica-Ihle (Rheumaklinik Berlin-Buch, Berlin), H Häntzschel (Medizinische Klinik und Poliklinik IV, Leipzig), G Hein (Friedrich Schiller Universität, Jena), S Mindt-Prüfert (Klinische Forschung Hamburg, Hamburg), JR Kalden (Medizinische Klinik III, Erlangen), T Karger (Rheumatologische Schwerpunktpraxis, Eduardus-Krankenhaus, Köln), A Krause (Immanuel-Krankenhaus, Berlin), K Krüger (Praxiszentrum St. Bonifatius, München), G Neeck (Biomedizinische Forschung und Entwicklung Rostock GmbH, Rostock), H Nüsslein (Krankenhaus Friedrichstadt, Dresden), S Wassenberg (Evangelisches Fachkrankenhaus GmbH, Ratingen), J Wollenhaupt (Allgemeines Krankenhaus Eilbeck, Hamburg), H Zeidler (Medizinische Hochschule, Hannover). Poland: J Szechinski, co-ordinating investigator for Poland (Katedra i Klinika Reumatologii i Chorob Wewnetrznych AM, Wroclaw), A Filipowicz-Sosnowska (Zaklad Reumatologii Szpital Kolejowy, Warszawa), S Jeka (Clinical Trials Office NZOZ ‘Nasz Lekarz’, Torun), EJ Kucharz (Katowice), S Mackiewicz (Klinika Reumatologii, Poznan), M Misterska-Skóra (NZOZ Materia Medica, Wroclaw), M Rell-Bakalarska (Przychodnia Przykliniczna Instytutu Reumatologii, Warszawa), L Szczepanski (Gabinety Profesorów, Lublin), S Sierakowski Department of Rheumatology Medical University, Bialystok), J Szerla (Krakowski Szpital Reumatologii i Rehabilitacja, Kraków), D Wierzbinska-Zarowny (Wojewodzki Zespol Rheumatologiczny, Sopot), I Zimmermann-Gorska (Katedra i Klinika Reumatologiczno Rehabilitacyjna i Chorob Wewnetrznych, Poznan).
-
Competing interests FB has received lecture fees and grant support from Merck Pharma GmbH and Nitec Pharma GmbH and is a consultant for these companies. GD was an employee of Merck KGaA and holds company stocks; she is also a consultant for NitecPharma AG, Reinach, Switzerland. AS and SW are employees of Nitec Pharma GmbH and hold company stocks. RA has received lecture fees from Merck Pharma GmbH and Nitec Pharma GmbH and is a consultant for these companies. JS, EG-I, SJ, KK and SS have no conflicts of interest.
-
Funding The study was supported by Merck KGaA, Darmstadt, Germany and NitecPharma AG, Reinach, Switzerland. Investigators were remunerated according to patient enrolment and suitable documentation. The study coordinator of Merck KGaA was involved in the analysis of the data and the reporting. The corresponding author had full access to all data at all times, had a major influence on the manuscript at all stages and had the final responsibility for the decision to submit for publication.
-
Ethics approval This study was conducted with the approval of the Ethikkommission der Charite, Berlin, Germany.
-
Provenance and peer review Not commissioned; externally peer reviewed.