Article Text

Abstract
Objective To assess safety, immunogenicity and efficacy in rheumatoid arthritis (RA) patients switched from long-term intravenous to subcutaneous (SC) abatacept.
Methods In this phase IIIb, open-label, single-arm trial, patients who completed ≥4 years of intravenous abatacept (in long-term extensions of two phase III studies) were enrolled to receive SC abatacept (125 mg/week). The primary objective was safety during the first 3 months after switching from intravenous therapy.
Results 123 patients entered the study (mean Disease Activity Score 28 (based on C reactive protein) and HAQ-DI of 3.4 and 0.94, respectively). At month 3, 120 (97.6%) patients were continuing to receive SC abatacept; no patients discontinued due to lack of efficacy. Adverse events (AEs) were reported in 49 (39.8%) patients through month 3. One patient (0.8%) discontinued due to an AE and one patient (0.8%) experienced a serious AE. Two (1.6%) patients had SC injection site reactions (erythema, pain), both with mild intensity. Clinical efficacy was maintained throughout. Limited impact on immunogenicity was observed when switching routes of administration.
Conclusion These data demonstrate that patients can switch from long-term monthly intravenous abatacept to a weekly fixed dose of 125 mg SC abatacept with no increased safety concerns. This study further supports SC abatacept as an alternative treatment option for patients with RA.
Statistics from Altmetric.com
Introduction
Abatacept, a T cell co-stimulation modulator, is approved for the treatment of moderate-to-severe rheumatoid arthritis (RA), initially as an intravenous formulation administered monthly according to a weight-tiered dosing regimen, and recently as a subcutaneous (SC) fixed dose. Clinically meaningful and sustained efficacy, along with consistent safety and tolerability, has been demonstrated in large numbers of patients treated with intravenous abatacept, including methotrexate (MTX)-naïve early RA patients up to 2 years,1 MTX-inadequate responders up to 7 years2 and antitumour necrosis factor (anti-TNF)-inadequate responders up to 4.5 years.3
For some patients, a switch from intravenous to a SC formulation is desirable. In a recent non-inferiority study, SC abatacept demonstrated comparable efficacy and safety to intravenous abatacept, including low immunogenicity and high patient retention.4 Other studies have verified that fixed-dose (125 mg/week) SC abatacept achieves stable serum concentrations above the target therapeutic Cmin of 10 µg/ml5 and that SC abatacept, administered as monotherapy or with MTX, in the absence of an intravenous loading dose, does not elicit significant immunogenicity.6 7 In addition, it has been demonstrated that temporary withdrawal and re-introduction of SC abatacept is well tolerated and has little impact on immunogenicity, efficacy and safety.8
Understanding the clinical impact of changing formulations in the months immediately following the switch is important with regard to safety, tolerability and efficacy. The phase IIIb ATTUNE (Abatacept in subjecTs who swiTch from intravenoUs to subcutaNeous thErapy) trial is the first study to evaluate these parameters in patients switching to SC abatacept from two intravenous abatacept clinical trials.9 10
Methods
Study design
ATTUNE was a 12-month, international, open-label, single-arm study. Patients (≥18 years) with active RA previously refractory to either MTX or anti-TNFs who had received ≥4 years of intravenous abatacept in AIM (Abatacept in Inadequate responders to Methotrexate study) or ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders) were eligible.3 9,–,11
The first dose of weekly SC abatacept (125 mg) was administered ≤30 days from the last intravenous dose (an intravenous abatacept dose was not administered on day 1). Changes to concomitant disease-modifying antirheumatic drugs, corticosteroids and/or non-steroidal anti-inflammatory drugs (NSAIDs) were not permitted until after month 3.
The primary objective was the assessment of safety through month 3. Immunogenicity through month 3 was a secondary endpoint. Efficacy end points were assessed throughout and are reported up to month 12; safety and immunogenicity were assessed and reported throughout.
Safety
Adverse events (AEs) and serious AEs (SAEs) classified using the Medical Dictionary for Regulatory Activities (MedDRA) were reported monthly. SC injection site reactions and autoimmune events were prespecified based on MedDRA preferred terms.
Immunogenicity
Serum samples were obtained prior to SC administration at each monthly visit through month 3, and quarterly thereafter. Immunogenicity to abatacept was recorded as the proportion of patients with a seropositive response (on-treatment or post-treatment (up to 85 days)) using an ELISA (used through month 3 as the primary immunoassay to facilitate comparisons with previous intravenous studies) or an electrochemiluminescence (ECL) immunoassay (higher sensitivity) used throughout (online supplementary text has further details).
Efficacy
Disease Activity Score 28 (DAS28), based on C reactive protein (CRP), was assessed monthly through month 3 and quarterly thereafter. Mean change from baseline and the proportions of patients in low disease activity state (DAS28 (CRP) ≤3.2) and remission (DAS28 (CRP) <2.6) were determined. Mean Health Assessment Questionnaire-Disability Index (HAQ-DI) scores over time were also assessed.
Statistical analyses
No formal statistical testing was performed. Safety, immunogenicity and efficacy were assessed for patients who received ≥1 dose of SC abatacept. Efficacy data are as observed.
Results
Patient disposition and baseline characteristics
In total, 123 patients from the AIM (n=71) or ATTAIN (n=52) trials were enrolled and treated in this study (online supplementary figure S1). Of these patients, 120 (97.6%) completed month 3 and 112 (91.1%) were continuing to receive SC abatacept at time of data analysis (median exposure 15.4 (range: 2–20) months).
Patients had mean DAS28 (CRP) of 3.4, mean HAQ-DI of 0.94, and mean tender and swollen joint counts of 8.9 and 4.8, respectively, at baseline. The mean (SD) interval between the last intravenous abatacept dose and first SC dose was 27.5 (6.2) days (range: 8–69 days); online supplementary table S1 and online supplementary text have additional details.
At baseline 99.2% of patients were receiving concomitant medication (disease-modifying antirheumatic drugs, NSAIDs or corticosteroids); mean (SD) corticosteroid dose was 2.4 (4.0) mg. After month 3, eight and seven patients added corticosteroids and NSAIDs, respectively, while two patients added MTX. The mean (SD) corticosteroid dose over the cumulative period was 2.8 (5.1) mg. Most patients (93.5%) received ≥13 months of SC abatacept.
Safety
Overall
Up to month 3, AEs were reported in 39.8% of patients (table 1); no individual AE occurred in ≥5%. One AE (musculoskeletal pain) led to discontinuation. Overall, 75.6% of patients experienced an AE during the cumulative period. One SAE (worsening of RA; moderate intensity, patient did not discontinue) occurred during the first 3 months. After month 3, 12 further SAEs occurred, three of which (breast cancer, sarcoidosis and brain neoplasm) resulted in discontinuation. No deaths were reported in the study or during follow-up (online supplementary text has further details on overall safety).
Safety summary
Infections reported up to month 3 (in >1 patient) were nasopharyngitis (four patients), urinary tract infection (three), bronchitis (two), gastroenteritis (two), sinusitis (two) and upper respiratory tract infection (two). No serious infections, malignancies or autoimmune events were reported during the first 3 months.
Serious infections, malignancies or autoimmune events occurring after month 3 were as follows: one serious infection (pneumonia; did not result in discontinuation), two malignancies (breast and uterine cancer, both SAEs subsequently resolved; patient with breast cancer discontinued) and two autoimmune events occurred (sarcoidosis (considered an SAE) and erythema nodosum (mild intensity)).
SC injection site reactions
Two SC injection site reactions occurred up to month 3, injection site erythema and injection site pain. Both events occurred following the first SC injection, were of mild intensity and not serious, and treatment continued. Erythema recurred following three subsequent injections, persisting for 4–5 days. No SC injection site reactions occurred after month 3.
Immunogenicity
Eight patients were seropositive based on ELISA through month 3 (table 2 and online supplementary text). Of these eight, six were already positive prior to enrolment. All eight seropositive patients continued treatment. At month 3, efficacy was maintained/improved in six of these patients; there was evidence of clinical worsening (increase in DAS28 (CRP) of >0.6 from baseline) in two patients. AEs experienced by these eight patients were generally not consistent with immune-mediated toxicities, except for one patient who developed sarcoidosis and discontinued from the study; none of the other seropositive patients discontinued from the study. None of these patients had an abatacept-induced seropositive result based on the ECL assay.
Immunogenicity summary
During the first 3 months of the study, no patient had an abatacept-induced seropositive result based on the ECL assay (table 2). After month 3, one patient demonstrated a seropositive result while on treatment, with a low titre of 10 (equal to assay cut-off value). This patient did not experience immune-mediated toxicity or SC injection site reactions; two AEs were reported by this patient during the study (eye infection and upper respiratory tract infection), neither of which were serious.
Of the eight patients who discontinued the study, none had seropositive results following withdrawal of therapy (assessed 28, 56 and 85 days after last dose).
Clinical efficacy
At baseline, mean (SD) DAS28 (CRP) and HAQ-DI scores in the overall population were 3.39 (1.26) and 0.94 (0.69), respectively. Improvements in disease activity and physical function achieved during intravenous treatment were maintained through month 12 of SC treatment (figure 1A,B). A considerable proportion of patients were in low disease activity state (43.4%) and DAS28 remission (32.0%) at baseline. These states were maintained through month 12 (figure 1C,D).

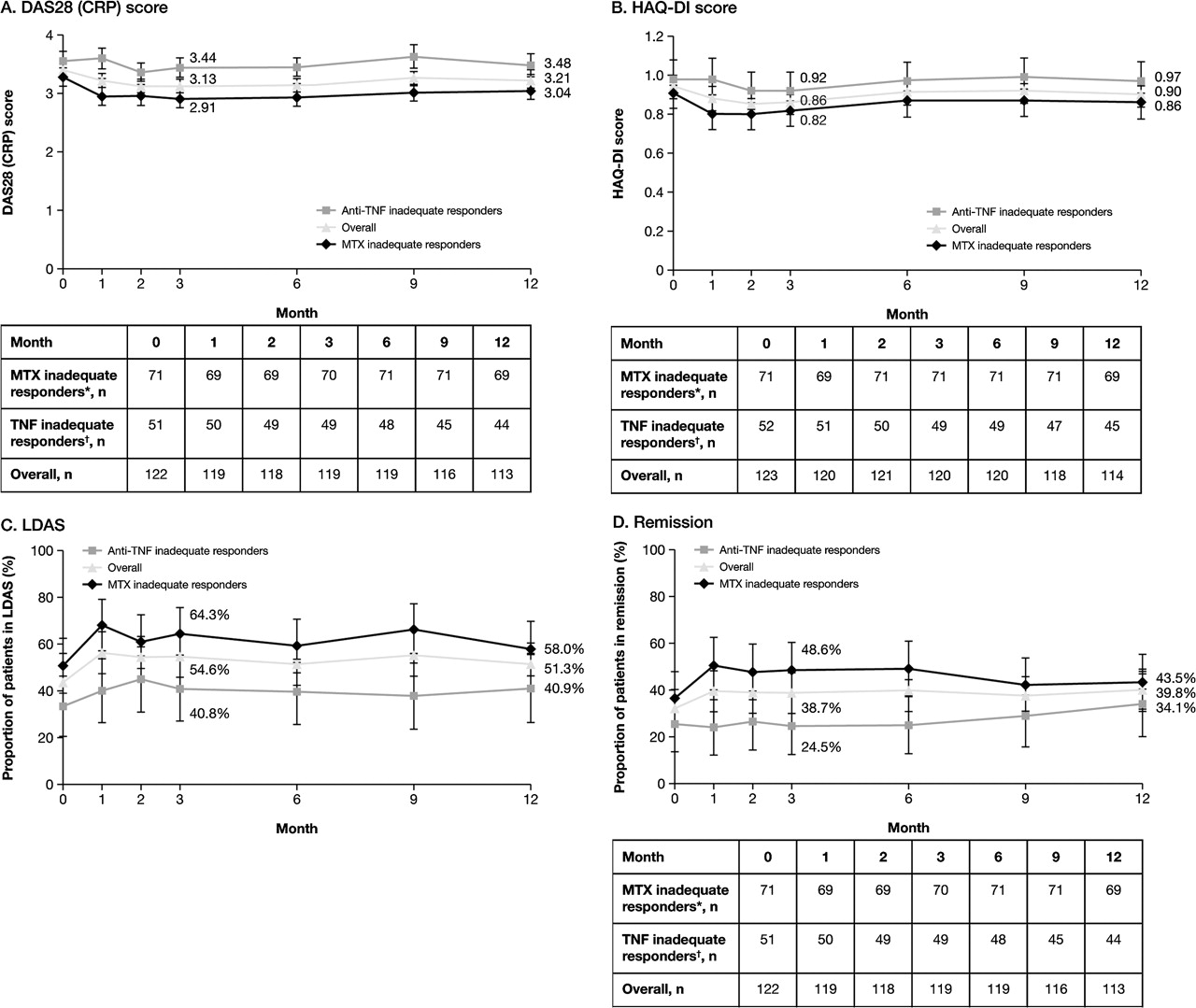{kind=link}
Maintenance of clinical efficacy. (A) Mean DAS28 (CRP) score over 1 year; error bars represent SE. (B) Mean HAQ-DI score over 1 year; error bars represent SE. (C) Proportion of patients sustaining LDAS over 1 year; error bars represent 95% CI. (D) Proportion of patients sustaining remission over 1 year; error bars represent 95% CI. LDAS is defined as DAS28 (CRP) ≤3.2 and remission as DAS28 (CRP) <2.6. *Participated in AIM; †Participated in ATTAIN. AIM, Abatacept in Inadequate responders to Methotrexate; ATTAIN, Abatacept Trial in Treatment of Anti-TNF INadequate responders; CRP, C reactive protein; DAS28 (CRP), Disease Activity Score 28 (based on C reactive protein); HAQ-DI, Health Assessment Questionnaire-Disability Index; LDAS, low disease activity state; MTX, methotrexate; TNF, tumour necrosis factor.
Discussion
The ATTUNE trial evaluated switching patients to 125 mg weekly SC abatacept from intravenous abatacept in the AIM and ATTAIN intravenous studies. Data presented demonstrate that switching from intravenous to SC administration was well tolerated, with a low risk of immunogenicity. The high retention rate (>90%) indicates that patients can be successfully transitioned from intravenous to SC abatacept, with few discontinuations for AEs or lack of efficacy. Mean disease activity and physical function scores achieved during long-term intravenous abatacept were maintained over 12 months of SC treatment; the ability to maintain efficacy benefits following a change in administration represents an important advance in our therapeutic armamentarium.
In the 3 months following the switch from intravenous to SC abatacept—which the authors feel is a clinically relevant timeframe to determine any safety events that may be associated with switching—the safety profile was consistent with other intravenous and SC abatacept studies.5 6 12 Treatment over the cumulative period did not result in an increased frequency of any known risk (including malignancies, autoimmune events and serious injections) or any additional risks, similar to findings from integrated analyses of intravenous13 and SC14 abatacept trials.
SC administration of some biologics can result in a relatively high rate of injection site reactions: 3.0–19.5% over 4–6 months.15,–,17 Here, injection site reactions were only reported in the first 3 months in 1.6% of patients (pain and erythema in only one patient each). All were mild, and no patient discontinued. In the pivotal ACQUIRE (Abatacept Comparison of sub(QU)cutaneous versus Intravenous in inadquate Responders to methotrexatE) phase IIIb SC versus intravenous abatacept study (n=1457), SC injection site reactions during the first 6 months were mostly mild and occurred in 2.6% of SC abatacept-treated patients versus 2.5% of intravenous abatacept-treated patients.4
Immunogenicity during biologic therapy has been associated with reduced efficacy and increased AEs.18,–,20 Rates of 43% are reported for infliximab over 1 year and 17–20% for adalimumab over 7 months.21,–,23 Rates from SC abatacept studies have been reported to be 2.0–3.0% over 3–4 months5 6 and <3.0% over 18 months.7 Here, the immunogenicity rate ranged from 0.8–6.6% (dependent on assay) over the first 3 months and cumulative period (median exposure 15.4 months), with no apparent impact on efficacy or safety outcomes. This is similar to a meta-analysis of long-term phase II and III intravenous abatacept studies, where rates of 2.8–3.0% were observed with no consistent pattern between immunogenicity and loss of efficacy or increased safety concerns.24 It should be noted that in our study, six of the eight seropositive patients (based on ELISA) were seropositive prior to enrolment. In addition, abatacept Cmin concentrations were consistent before and after demonstration of the positive response, suggesting that immunogenicity did not affect systemic drug exposure (data not presented).
This study should be interpreted within its limitations. The open-label nature of the study and the relatively small patient numbers should be considered. As efficacy analyses use as-observed data, findings are dependent on patient retention. However, relatively few patients discontinued throughout (<10%).
In summary, ATTUNE demonstrates that patients can switch from long-term monthly weight-tiered intravenous to weekly fixed dose SC abatacept, with no increased safety concerns or risk of immunogenicity, and with maintained efficacy. Taken together with other recent studies,4 7 8 these data further support SC abatacept as an alternative treatment option for patients with RA.
Acknowledgments
The authors would like to thank Róisín O'Connor, Medicus International, for her editorial support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding This study was sponsored, and editorial support funded, by Bristol-Myers Squibb.
-
Competing interests ECK has received consulting fees from Abbott Laboratories', AstraZeneca Pharma, Biotest, Bristol-Myers Squibb Company, Centocor, Inc, F Hoffmann-La Roche Inc, Genentech Inc, Merck, Nycomed, Pfizer Pharmaceuticals and UCB; has received research grants from Abbott Laboratories, Amgen Inc, AstraZeneca Pharmaceuticals LP, Bristol-Myers Squibb, Centocor, Inc, F Hoffmann-LaRoche Inc, Genzyme, Merck, Novartis Pharmaceuticals, Pfizer Pharmaceuticals and UCB; and has participated in speakers' bureau for Abbott Laboratories, Bristol-Myers Squibb, Hoffmann-La Roche Inc, Merck, Pfizer Pharmaceuticals and UCB. JMK has received consulting fees from Abbott Laboratories, Amgen Inc, Bristol-Myers Squibb, Centocor Ortho Biotech Inc, Genentech, Genentech and Biogen IDEC Inc, Merck Pharmaceuticals, Ortho Biotech Products L P, Pfizer Inc, Roche and UCB, Inc; has received research grants from Abbott Laboratories, Amgen Inc, Bristol-Myers Squibb, Centocor Ortho Biotech Inc, Genentech, Genentech and Biogen IDEC Inc, Human Genome Sciences, Inc, Merck Pharmaceuticals, Ortho Biotech Products LP, Pfizer Inc, Roche, Roche Diagnostics and UCB, Inc; has participated in speakers' bureau for Genentech, Genentech and Biogen IDEC Inc, and Roche; and has stock holdings in CORRONA. AR has received consulting fees and honoraria from Bristol-Myers Squibb, GlaxoSmithKline and Pfizer Inc, and has participated in speakers' bureau for Bristol-Myers Squibb, GlaxoSmithKline and Pfizer Inc. JB has received consulting fees from Bristol-Myers Squibb, has participated in a speakers' bureau for Bristol-Myers Squibb, and is the owner of Box Arthritis and Rheumatology of the Carolinas PLLC. CA-M and MGE have nothing to disclose. ML has received research grants from Bristol-Myers Squibb. AL, ID and RS are full-time employees of, and stock-holders in, Bristol-Myers Squibb. RA and SG were full-time employees of, and stock-holders in, Bristol-Myers Squibb, when this study was carried out.
-
Patient consent Freely given written informed consent was obtained from each subject, or in those situations where consent could not be given by the subject, their legally acceptable representatives, at the final quarterly dosing visit for AIM or ATTAIN prior to participation in ATTUNE, including informed consent for any screening procedures conducted to establish subject eligibility in the study
-
Provenance and peer review Not commissioned; externally peer reviewed