Article Text

Abstract
Objective To evaluate efficacy of tocilizumab in US patients with moderate to severe active rheumatoid arthritis (RA) and inadequate clinical response to disease-modifying antirheumatic drugs (DMARD). Safety-related outcomes were also analysed.
Methods The rapid onset and systemic efficacy study was a 24-week, randomised, double-blind trial. Patients were randomly assigned 2:1 to tocilizumab 8 mg/kg (n=412) or placebo (n=207) every 4 weeks while continuing background DMARD in both groups.
Results The primary efficacy endpoint, percentage of patients achieving ACR50 response at week 24, was higher with tocilizumab versus placebo (30.1% vs 11.2%; p<0.0001). Percentages of ACR20 and ACR50 responders were significantly higher with tocilizumab versus placebo as early as week 4 and continued to week 24; more patients in the tocilizumab versus placebo group also achieved ACR70 responses beginning at week 8 (p<0.01). Significant improvements associated with tocilizumab versus placebo were seen in routine assessment of patient index data responses, EULAR good response, DAS28 and percentages of patients achieving low disease activity and clinical remission (based on DAS28). A substudy examining early response to therapy showed improved patient global assessment of disease activity (p=0.005) and pain (p=0.01) and DAS28 (p=0.007) with tocilizumab versus placebo at day 7. Safety findings were consistent with the known tocilizumab safety profile; rates of serious infections (per 100 patient-years) were 7.87 (95% CI 4.30 to 13.2) and 1.20 (95% CI 0.03 to 6.66) in the tocilizumab and placebo groups, respectively.
Conclusions This study demonstrated the efficacy of tocilizumab in improving measures of disease activity in patients with RA who failed to respond adequately to DMARD therapy. Rapid improvement in clinical outcomes was demonstrated in a substudy as early as week 1 as shown by DAS28 scores, patient measures and C-reactive protein.
Trial Registry no NCT00531817
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Early and aggressive treatment of rheumatoid arthritis (RA) is associated with improved outcomes, including lower disease activity, improved physical function and quality of life and reduced radiographic disease progression.1,–,6 Despite advances in how we treat RA,7 ,8 many patients still fail to respond or respond only partly to disease-modifying antirheumatic drugs (DMARD) or anti-tumour necrosis factor (TNF) therapy.9
Tocilizumab is a humanised, monoclonal, anti-interleukin (IL) 6 receptor antibody that binds to both soluble and membrane-bound IL-6 receptor, inhibiting IL-6R signal transduction to inflammatory mediators.10 The efficacy and tolerability of tocilizumab have been demonstrated previously in combination with traditional DMARD in patients with inadequate responses to earlier DMARD9 ,11 or anti-TNF biological agents,12 and as monotherapy in comparison with methotrexate.13
The aim of the current study was to evaluate further the efficacy and tolerability of tocilizumab for 6 months for the first time in a US patient population with moderate to severe active RA who had an inadequate clinical response to DMARD. Unlike previous studies of tocilizumab in DMARD inadequate response patients,9 ,11 this study also assessed the response to therapy in the first week after the first infusion for a subset of study participants. In addition, this study employed as a primary endpoint the highest threshold of the clinically relevant outcome (American College of Rheumatology (ACR) 50% improvement; ACR50).
Patients and methods
Study design and patient population
The Rapid Onset and Systemic Efficacy (ROSE) study was a 24-week, randomised, double-blind, placebo-controlled, parallel-group, multicentre phase IIIb clinical trial. According to revised 1987 ACR criteria, this study included adults diagnosed with active RA for at least 6 months who were experiencing an inadequate clinical response to DMARD as determined by the investigator. Eligible patients had six or more swollen joints and six or more tender joints at screening and baseline, and either a C-reactive protein (CRP) level of 95.24 nmol/l or greater or an erythrocyte sedimentation rate (ESR) of 28 mm/h or greater at screening (normal reference ranges are 0.76–28.5 nmol/l for CRP and 0–25 mm/h for ESR).14 Patients were randomly assigned 2:1 to receive tocilizumab 8 mg/kg intravenously every 4 weeks plus stable antirheumatic therapy including DMARD (tocilizumab group) or placebo intravenously every 4 weeks plus DMARD (placebo group). Permitted DMARD (at stable doses ≥7 weeks before study baseline) included methotrexate, chloroquine, hydroxychloroquine, parenteral gold, sulfasalazine, azathioprine and leflunomide. Doses of these permitted DMARD (in addition to corticosteroid and non-steroidal anti-inflammatory drug doses) were required to remain stable throughout the study; however, dose reductions were allowed as clinically warranted for safety reasons. Patients were required to discontinue previous biological therapy (eg, adalimumab, etanercept, infliximab, abatacept, anakinra, rituximab) before randomisation. Rescue therapy (two courses of tocilizumab 8 mg/kg every 4 weeks, in place of the assigned study drug) was permitted from week 16 for patients who did not achieve a 20% or greater improvement from baseline in swollen and tender joint counts.
A subset of patients with CRP levels of 95.24 nmol/l or greater who received one or more administration of study drug and who agreed to participate in an ancillary substudy had additional assessments on days 3 and 7 to measure early response on disease activity control. The analysis for the substudy was predefined in the protocol and was not to alter the enrolment or design of the study. It was performed by an independent data review committee. The study management team, investigational staff and monitors remained blinded to individual patients' treatment assignments.
All patients gave written informed consent, and the study complied with the principles of good clinical practice and the Declaration of Helsinki.
Assessments
The primary efficacy endpoint was the ACR50 response at week 24. In addition, the following were determined every 4 weeks to week 24: ACR20/50/70 responses;15 ,16 European League Against Rheumatism (EULAR) responses;16,–,18 disease activity score in 28 joints (DAS28),19 including proportions of patients with clinically meaningful improvement (change from baseline in DAS28 of ≥1.2) and patients achieving low disease activity (LDA, DAS28 ≤3.2) or clinical remission (DAS28 <2.6);18 ESR and CRP levels; functional assessment of chronic illness therapy—fatigue (FACIT-F);20 and routine assessment of patient index data (RAPID3) scores (on a scale of 0–10) derived from the multidimensional health assessment questionnaire (MDHAQ).21 In the substudy, additional efficacy assessments for the early response on disease activity control included CRP levels on days 3 and 7; joint counts, patient and investigator global assessment of disease activity, patient pain assessment, MDHAQ and ESR on day 7. Safety assessments performed throughout the 24-week treatment period included adverse event (AE) reporting, withdrawals from the study, deaths, serious infections, malignancies and laboratory parameters, such as liver enzyme levels, neutrophils, platelets and lipids.
Statistical analysis
The intent-to-treat (ITT) population consisted of all randomly assigned patients who received at least one administration of study medication. The safety population included all patients who received at least one administration of study medication and who had at least one safety assessment after receiving study medication.
Patients who received rescue therapy and patients who did not have data required to assess efficacy outcomes at week 24 were classified as non-responders. Last observation carried forward methodology was used for missing joint count data. A two-sided Fisher's exact test was used to determine p values for ACR responses, LDA and DAS28 remission differences between groups at each time point. The Cochrane–Mantel–Haenszel χ2 test was used to determine p values for EULAR response between groups at each time point. For continuous variables, such as DAS28, RAPID3, FACIT-F, CRP and ESR, p values and 95% CI for the difference of change from baseline between treatment groups were obtained using an analysis of covariance model with baseline as a covariate and treatment group as a factor. Groups were not stratified by history of TNF inhibitor use.
Results
Patients
Patient disposition is shown in figure 1. The ITT population included 614 patients, 409 and 205 in the tocilizumab and placebo groups, respectively. Baseline demographics and disease characteristics were similar between groups (table 1). Overall, most patients were women and were white, with a mean age of approximately 55 years and a mean duration of RA of 8.5 years. More than 25% of patients had had RA for at least 10 years. The mean DAS28 was 6.5 at baseline in both treatment groups. Most patients in both treatment groups reported using one background DMARD at baseline. The mean number of previous DMARD or anti-TNF was 1.4 and 1.3 in the tocilizumab and placebo groups, respectively. The proportions of patients who discontinued previous treatment with anti-TNF or non-biological DMARD were similar between groups, as was the proportion of patients currently receiving methotrexate as active DMARD (86.3% tocilizumab, 86.8% placebo). Hypertension was the most common comorbid diagnosis in these patients.

Patient disposition. DMARD, disease-modifying antirheumatic drug; ITT, intent-to-treat.
Baseline demographics and disease characteristics (ITT population)
Efficacy assessments
The primary efficacy endpoint, the percentage of patients achieving an ACR50 response at week 24, was significantly higher in the tocilizumab group than in the placebo group (30.1% vs 11.2%; p<0.0001). As shown in figure 2, percentages of both (A) ACR20 and (B) ACR50 responders were significantly higher in the tocilizumab versus the placebo group as early as week 4 and continued to week 24; significantly more patients in the tocilizumab versus the placebo group also achieved (C) ACR70 responses at week 8 and at all subsequent time points.

Percentages of patients achieving (A) ACR20, (B) ACR50 and (C) ACR70 responses over time. ACR20: *p<0.0001. ACR50: *p=0.0002, †p<0.0001, ‡p=0.0017, §p=0.0015. ACR70: *p=0.0002, †p=0.0076, ‡p<0.0001. All comparisons were tocilizumab plus DMARD vs placebo plus DMARD. ACR, American College of Rheumatology; DMARD, disease-modifying antirheumatic drug; TCZ, tocilizumab.
Proportions of patients achieving a EULAR good response increased over time in the tocilizumab group and were significantly higher than in the placebo group at all time points starting at week 4 (13.2% vs 2.0% of patients in the tocilizumab vs placebo groups; p<0.0001 for between-group comparison across response categories). At week 24, 32.5% of patients in the tocilizumab group versus 5.9% of patients in the placebo group achieved a EULAR good response (p<0.0001).
The mean DAS28 score decreased from baseline to week 24 in both treatment groups starting at week 4, but the adjusted mean change from baseline was significantly greater in the tocilizumab versus the placebo group at all time points (p<0.0001, figure 3A). In the tocilizumab group, the mean DAS28 decreased from 6.53 at baseline to 3.24 at week 24. The proportion of patients achieving a clinically meaningful decrease in DAS28 (≥1.2 points from baseline) was significantly higher in the tocilizumab versus the placebo group at all time points from week 4 onwards (figure 3B; 87.9% in the tocilizumab group vs 53.4% in the placebo group at week 24, p<0.0001). Furthermore, significantly greater proportions of patients in the tocilizumab versus the placebo group achieved both LDA and clinical remission at all time points from week 4 onwards (figure 3C,D). At week 24, 50.7% of patients achieved LDA and 38.4% reached clinical remission in the tocilizumab group (p<0.0001 vs placebo for both measures).

(A) Mean DAS28; (B) percentages of patients achieving clinically meaningful improvement (DAS28 improvement from baseline ≥1.2); (C) LDA (DAS28≤3.2); and (D) clinical remission (DAS28<2.6) over time. *p<0.0001, tocilizumab plus DMARD vs placebo plus DMARD. Panel A: p values compare adjusted mean changes from baseline between the two treatment groups. DAS28, disease activity score in 28 joints; DMARD, disease-modifying antirheumatic drug; LDA, low disease activity; TCZ, tocilizumab.
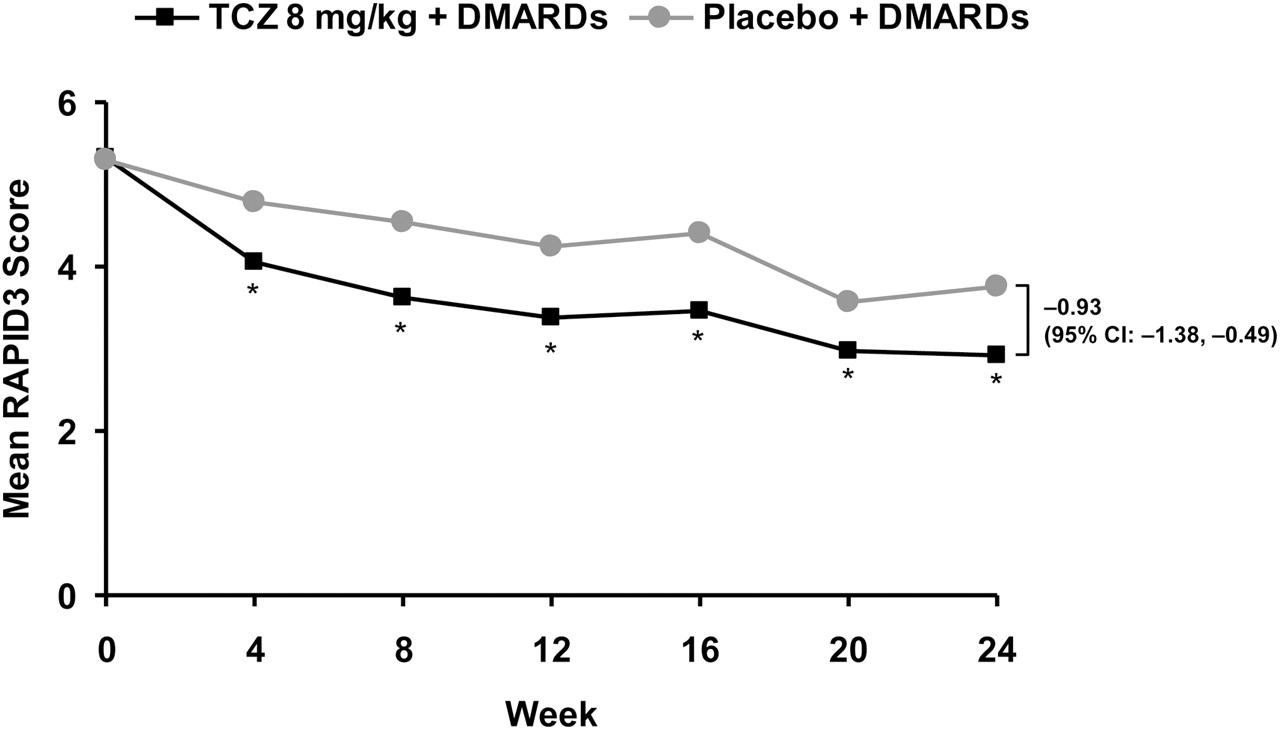
Mean RAPID3 scores decreased during the first week of treatment in both groups; improvements were significantly greater for patients in the tocilizumab versus the placebo group on days 3–7 (day 7 mean change from baseline, −1.00 vs −0.51 for tocilizumab vs placebo, respectively; difference in adjusted mean change, −0.47, 95% CI −0.78 to −0.16; p=0.0029).
(Note: Please see supplementary appendix, available online only, for additional information on RAPID3 scores during the first week of treatment.)
Significantly greater improvements from baseline in the tocilizumab versus the placebo group were also seen at week 4 and at all subsequent time points (figure 4). By week 24, the mean change from baseline in the RAPID3 score was −2.33 in the tocilizumab group versus −1.29 in the placebo group (p<0.0001).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean RAPID3 score over time. *p<0.0001, †p=0.0011; adjusted mean change from baseline for tocilizumab plus DMARD vs placebo plus DMARD. DMARD, disease-modifying antirheumatic drug; RAPID3, routine assessment of patient index data; TCZ, tocilizumab.
Mean CRP levels were significantly lower in the tocilizumab versus the placebo group starting at week 4 (39.05 vs 162.86 nmol/l) and remained significantly lower at all time points (p<0.0001, data available in supplementary appendix figure S5, available online only). Mean ESR showed a significantly greater mean decrease from baseline in the tocilizumab versus the placebo group starting at week 4 (−29.82 vs −3.52 mm/h; p<0.0001), which was sustained to week 24 (−34.72 vs −5.70 mm/h; p<0.0001). Improvement in mean FACIT-F scores was evident by week 8 in the tocilizumab group versus the placebo group and was sustained to week 24 (p<0.05). At week 24, mean change from baseline was 8.43 versus 5.89 in the tocilizumab group versus the placebo group, respectively; the difference in adjusted mean change from baseline at week 24 was 2.73 (95% CI 0.45 to 5.00; p=0.0188).
Although RAPID3 scores decreased during the first week of treatment in both groups, improvements were significantly greater for patients in the tocilizumab versus the placebo group on days 3–7 (day 7 mean change from baseline, −1.00 vs −0.51 for tocilizumab vs placebo, respectively; difference in adjusted mean change −0.47, 95% CI −0.78 to −0.16; p=0.0029).
Early disease activity control substudy
This substudy included 40 patients in the tocilizumab group and 22 in the placebo group (see supplementary appendix, available online only, for baseline demographics and clinical characteristics). Several measures showed significant differences by day 7 of treatment. At day 7, CRP levels had decreased significantly more from baseline in the tocilizumab versus the placebo group (−256.2 vs −29.5 nmol/l; p<0.001), as did the ESR (−25.6 vs 0.30 mm/h; p<0.001). In addition, both patients' global assessment of disease activity (−16.2 vs 0.8; p=0.005) and assessment of pain (−12.2 vs 1.4; p=0.01) improved significantly more in the tocilizumab versus the placebo group; DAS28 (−1.16 vs −0.27; p=0.007) also improved significantly more in the tocilizumab group. Statistically significant differences between the tocilizumab and placebo groups were not observed at day 7 for tender and swollen joint counts or physician's global assessment of disease activity (table 2). Of note, no significant changes from baseline in the number of DMARD, methotrexate dose, or concomitant oral steroid dose were observed.
Results of the early disease activity control substudy (N=62): change from baseline to week 1
Safety
Table 3 presents an overview of AE during double-blind treatment. Serious AE were reported in 7.3% of patients in the tocilizumab group and 5.4% of patients in the placebo group, resulting in serious AE rates (per 100 patient-years) of 24.16 (95% CI 17.48 to 32.54) and 19.41 (95% CI 10.94 to 31.07), respectively. Twenty-seven patients (6.6%) withdrew from the treatment because of AE in the tocilizumab group versus eight patients (3.9%) in the placebo group. The most common AE leading to treatment withdrawal in the tocilizumab group were cellulitis (n=2), staphylococcal infection (n=2) and abnormal liver function tests (n=2). Three deaths were reported during the study, all of which occurred in the tocilizumab group: sepsis, haemorrhagic stroke and coronary artery arteriosclerosis. The deaths due to sepsis and haemorrhagic stroke were considered by the investigator to be possibly related to study treatment. The death due to coronary artery arteriosclerosis was considered unrelated to treatment.
Overview of AE during double-blind treatment (safety population)
Overall AE rates (per 100 patient-years) were 472.52 (95% CI 441.12 to 505.56) and 330.09 (95% CI 292.29 to 371.42) in the tocilizumab and placebo groups, respectively. The most common AE, occurring in over 5% of patients, were upper respiratory tract infection (7.6% tocilizumab, 4.9% placebo), urinary tract infection (5.9% tocilizumab, 5.4% placebo), diarrhoea (6.1% tocilizumab, 4.9% placebo) and worsening of RA (2.2% tocilizumab, 8.3% placebo).
Serious infections, gastrointestinal perforations and neoplasms
Twelve patients in the tocilizumab group and one patient in the placebo group had serious infections during double-blind treatment, representing a rate per 100 patient-years of 7.87 (95% CI 4.30 to 13.2) and 1.20 (95% CI 0.03 to 6.66), respectively. In the tocilizumab group, the infections were most commonly cellulitis (three patients, 0.7%) and pneumonia (three patients, 0.7%). Time to serious infections in the tocilizumab group ranged from day 32 to day 170 after the start of therapy (see supplementary appendix table S4, available online only). No opportunistic infections, including tuberculosis, were reported during the study, and no cases of gastrointestinal perforation. Seven neoplasms were reported, four in the tocilizumab group and three in the placebo group. In the tocilizumab group, the neoplasms were one malignant neoplasm of the eye (right temporal conjunctival intraepithelial neoplasia, carcinoma in situ, reported on day 146 from first infusion) and one lipoma of the cecum (not malignant, day 97), both of which were reported as serious AE, as well as one basal cell carcinoma (day 106) and one skin neoplasm (day 112), which were not reported as serious AE. In the placebo group, there was one report of non-melanoma skin cancer (squamous cell carcinoma of left cheek in situ, day 116); one diffuse large B-cell lymphoma (day 170) and one renal cell carcinoma (day 40). All three neoplasms in the placebo group were reported as serious AE.
Laboratory parameters
In the tocilizumab group, mean ALT, AST and total bilirubin levels increased by week 4 and generally remained elevated to week 24, but remained within normal ranges. Shifts in ALT and AST from normal at baseline to greater than the upper limit of normal (ULN) occurred in more patients in the tocilizumab group than in the placebo group: in the tocilizumab group, shifts in ALT up to 1.5× ULN occurred in 23.5% of patients; shifts to greater than 1.5× to 2× ULN occurred in 12.7%, and shifts to greater than 2× to 3× ULN occurred in 5.0% (vs 11.1%, 2.2% and 1.7%, respectively, in the placebo group). In addition, 10 patients (2.6%) had shifts to greater than 3× to 5× ULN and two patients (0.5%) had shifts to greater than 5× ULN in the tocilizumab group, compared with one patient (0.6%) in each category for the placebo group. Shifts in AST from normal at baseline to up to 1.5× ULN, greater than 1.5× to 2× ULN, greater than 2× to 3× ULN and greater than 3× to 5× ULN occurred in 26%, 4.9%, 2.3% and 0.8% of patients, respectively, in the tocilizumab group (vs 9.6%, 1.6%, 1.1% and 0.5%, respectively, in the placebo group). No patients in either group had shifts in AST to greater than 5× ULN (see supplementary appendix table S5, available online only). Most liver enzyme elevations were single occurrences with no cases of drug-induced injury.
The mean neutrophil count decreased in the tocilizumab group from 5.73×109/l at baseline to 4.27×109/l at week 4 and 3.75×109/l at week 24, while there was little change over time in the placebo group. In the tocilizumab group, 27.9% of patients had neutrophil levels less than the lower limit of normal (1.96×109/l) at some point during the study versus 4.0% of patients in the placebo group. Twelve (2.9%) patients in the tocilizumab group had common toxicity criteria (CTC) grade 3 neutropenia (0.5 to <1.0×109/l); none had grade 4 neutropenia. There was no apparent relationship between low neutrophil counts and the occurrence of infections. No serious infections were associated with a neutrophil count less than 1.0×109/l within 2 weeks of infection. The mean platelet count also decreased in the tocilizumab group, from 329.1×109/l at baseline to 242.0×109/l at week 4 and 241.6×109/l at week 24, while the placebo group showed little change over time. Thirty (7.4%) patients experienced CTC grade 1 platelet levels (75.0×103/μl to below lower limit)22 in the tocilizumab group versus none in the placebo group. No platelet level decreases to CTC grades 2–4 were reported in either group, and there were no reports of bleeding events associated with grade 1 platelet count.
Mean values of total cholesterol, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and triglycerides increased in the tocilizumab group from baseline to the end of the study, while the placebo group showed little change over time. Most of the increase was within the first 4–8 weeks of therapy and reached a plateau by weeks 12–16 (see supplementary appendix table S6, available online only). Individual patient shifts in lipid parameters from normal levels at baseline to levels above adult treatment panel III recommended thresholds23 at the end of the study are also shown in the supplementary appendix (table S6), available online only.
Discussion
At the time this study was designed, tocilizumab was not yet approved in the USA for the treatment of active RA nor for any other disorder; therefore, an active placebo design (ie, in which patients remained on their current medications and were either unable to or chose not to combine therapy with an older biological agent) was employed. The addition of tocilizumab to the treatment regimen of patients with moderate to severe active RA, who previously demonstrated an incomplete response to traditional DMARD and/or anti-TNF, resulted in significantly improved ACR50 response rates versus placebo; tocilizumab treatment was also associated with significant improvements in ACR20 and ACR70 response rates and DAS28 and RAPID3 scores. The benefits of tocilizumab therapy were seen as early as week 4, and were sustained to 24 weeks of treatment. Furthermore, the substudy evaluating early response on disease control suggested that several clinically relevant and statistically significant improvements, including patients' global assessments of disease activity and pain, DAS28, and reduction of CRP level and ESR, were observed within 7 days after the first infusion of tocilizumab. At 1 week, while patient-reported measures were significantly improved, physician-reported measures of global disease activity, tender and swollen joint counts were not. It may be important to realise the improvement in patient-reported outcomes, early in treatment compared with physician measures, when disease activity is measured.
Previous studies have shown that physician measures have a higher rate of placebo response, and are among the least efficient measures in clinical trials when compared with patient-reported outcomes.24 ,25 Our study provides additional evidence suggesting that patients are probably able to identify improvements more accurately and earlier than physicians. We believe this is an important finding to consider when designing and interpreting outcomes of randomised clinical trials.
Safety findings in this study were consistent with the known safety profile of tocilizumab. The rate of serious infection was higher in tocilizumab-treated patients (7.87 vs 1.20), although there were no occurrences of opportunistic infections—including tuberculosis—reported during the trial. Four neoplasms were reported in the tocilizumab group and three in the placebo group; there were no cases of gastrointestinal perforations in either arm.
There was no clinical evidence of liver injury or clinical hepatitis following treatment with tocilizumab. Mean lipid levels increased in patients treated with tocilizumab compared with placebo, but the clinical relevance of these lipid elevations is unclear in the RA population at this time.
While a number of clinical trials have been conducted in studying the efficacy and safety of tocilizumab with DMARD, this is the first report of efficacy and safety data in a US population. In addition, we attempted to identify a population in routine care who may be treated with not only methotrexate but other DMARD, with the subsequent addition of tocilizumab to improve outcomes given an inadequate response with previous therapy. The substudy group with disease activity measurements very early at 1 week after treatment is also unique and provides useful information for practising rheumatologists about the time to treatment response. As in other randomised clinical trials, the study has similar limitations including restrictions on patient enrolment, lack of major comorbidities represented among the treated patients, limited use of additional medications and strict dosing regimens. These limitations may potentially have an impact on the results.
In conclusion, these data suggest tocilizumab, an anti-IL-6R monoclonal antibody, has early and sustained efficacy in patients with RA who have failed to respond adequately to previous DMARD therapy.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
{kind=link}
Footnotes
-
Funding This study was funded by Roche. Support for third-party writing assistance for this manuscript, furnished by Marci Mikesell, PhD, was provided by Embryon. Third-party writing assistance for this paper was provided by F Hoffmann-La Roche.
-
Competing interests YY has received research grants and consulting fees from Genentech, a member of the Roche Group. He has received consulting fees from Bristol-Myers Squibb, Celgene, Centocor and UCB and has ownership/partnership in UCB. YY is a speaker for Bristol-Myers Squibb and Pfizer. JRC has received research grants and consulting fees from Amgen, Centocor, Corrona, Abbott, Roche and UCB. AI is a speaker for Amgen and Wyeth Pharmaceuticals. HB has received research grants and consulting fees from Roche. He is also a speaker for Roche. RLM is an employee of Genentech. LLT is a consultant of Roche. AK has received research grants and consulting fees from Roche.
-
Ethics approval This study was conducted with the approval of the ROSE protocol and study design were approved by a central institutional review board and by individual institutional review boards.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.