Article Text

Abstract
BACKGROUND Systemic sclerosis (SSc, scleroderma) in either its diffuse or limited skin forms has a high mortality when vital organs are affected. No treatment has been shown to influence the outcome or significantly affect the skin score, though many forms of immunosuppression have been tried. Recent developments in haemopoietic stem cell transplantation (HSCT) have allowed the application of profound immunosuppression followed by HSCT, or rescue, to autoimmune diseases such as SSc.
METHODS Results for 41 patients included in continuing multicentre open phase I/II studies using HSCT in the treatment of poor prognosis SSc are reported. Thirty seven patients had a predominantly diffuse skin form of the disease and four the limited form, with some clinical overlap. Median age was 41 years with a 5:1 female to male ratio. The skin score was >50% of maximum in 20/33 (61%) patients, with some lung disease attributable to SSc in 28/37 (76%), the forced vital capacity being <70% of the predicted value in 18/36 (50%). Pulmonary hypertension was described in 7/37 (19%) patients and renal disease in 5/37 (14%). The Scl-70 antibody was positive in 18/32 (56%) and the anticentromere antibody in 10% of evaluable patients. Peripheral blood stem cell mobilisation was performed with cyclophosphamide or granulocyte colony stimulating factor, alone or in combination. Thirty eight patients had ex vivo CD34 stem cell selection, with additional T cell depletion in seven. Seven conditioning regimens were used, but six of these used haemoimmunoablative doses of cyclophosphamide +/- anti-thymocyte globulin +/- total body irradiation. The median duration of follow up was 12 months (3–55).
RESULTS An improvement in skin score of >25% after transplantation occurred in 20/29 (69%) evaluable patients, and deterioration in 2/29 (7%). Lung function did not change significantly after transplantation. One of five renal cases deteriorated but with no new occurrences of renal disease after HSCT, and the pulmonary hypertension did not progress in the evaluable cases. Disease progression was seen in 7/37 (19%) patients after HSCT with a median period of 67 (range 49–255) days. Eleven (27%) patients had died at census and seven (17%) deaths were considered to be related to the procedure (direct organ toxicity in four, haemorrhage in two, and infection/neutropenic fever in one). The cumulative probability of survival at one year was 73% (95% CI 58 to 88) by Kaplan-Meier analysis.
CONCLUSION Despite a higher procedure related mortality rate from HSCT in SSc compared with patients with breast cancer and non-Hodgkin's lymphoma, the marked impact on skin score, a surrogate marker of mortality, the trend towards stabilisation of lung involvement, and lack of other treatment alternatives justify further carefully designed studies. If future trials incorporate inclusion and exclusion criteria based on this preliminary experience, the predicted procedure related mortality should be around 10%.
- stem cells
- scleroderma
- systemic sclerosis
- bone marrow transplantation
Statistics from Altmetric.com
Progressive systemic sclerosis (SSc) is a heterogeneous condition characterised by the deposition of excess collagen in skin and internal organs. Limited cutaneous and diffuse cutaneous forms of the disease are distinguishable not only by the extent of skin involvement but also by autoantibody profile, the pattern of organ involvement, and the additional cutaneous manifestations of limited disease (formerly referred to as the CREST syndrome). Both forms of the disease are associated with vascular abnormalities clinically manifest as Raynaud's phenomenon.
Numerous lines of evidence suggest that an inflammatory process with an autoimmune component precedes the development of fibrosis. Antibodies to nuclear proteins are commonly present before the development of disease manifestations other than Raynaud's phenomenon.1Reactivity to the nuclear autoantigens, topoisomerase I and the CEN proteins, are rarely seen other than with this disease and are associated with particular HLA-D genotypes.2 An inflammatory response with lymphocytic infiltration is evident early in the disease, and is well documented in skin3 and lung.4-7 Furthermore, the similarity of the condition to aspects of graft versus host disease has been frequently noted.8 ,9 This suggests that immune allo-reactivity can induce excessive fibrosis histologically similar to that seen in SSc. An increased frequency of microchimerism by haemopoietic cells derived from offspring has been reported in patients with SSc,10 ,11 and this could drive allogeneic immune responses analogous to graft versus host disease. Immunological involvement in the pathogenesis of the disease is supported by the clear thymic and T cell abnormalities found before the development of disease in the UC Davis spontaneous chicken model of scleroderma.12-14 Despite this evidence, the relation between autoimmune responses and the vascular pathology is unclear.15 Vascular abnormalities may be evident many years before the onset of disease.16 Similarly, the extent to which autoimmune responses and inflammation contribute to the maintenance of fibrosis remains unresolved. The perceived failure of immunosuppressive treatments to reverse established fibrosis suggests that, once initiated, the fibrotic process becomes independent of the immune drive and continues as an autonomous process.
Severe forms of the disease, and rapidly progressive diffuse SSc in particular, are associated with a significant mortality secondary to pulmonary, cardiac, renal, and gastrointestinal disease. Recent data suggest an overall five year mortality of 30%, with subgroup differences of up to 58%, depending on the clinical features at presentation.17 No treatment has proved effective in preventing disease progression or reversing fibrosis. A blinded randomised clinical trial of d-penicillamine in high and very low dose (125 mg on alternate days) produced the same results for skin thickness, which was considered by the authors probably to be similar to the natural history.18 α Interferon did not demonstrate a clinically significant effect,19 and low dose oral methotrexate showed conflicting results.20 Other studies of immunosuppressive regimens are continuing.
Although the limited cutaneous form of the disease has a much better overall prognosis, some patients in this group develop potentially fatal pulmonary fibrosis or primary pulmonary hypertension. This latter complication of disease has been reported in a few cases to respond to the administration of vasodilators, immunosuppression, and anticoagulation.21
Development of autologous stem cell transplantation for autoimmune disease
Autologous haemopoietic stem cell transplantation (HSCT) has been developed as a potential treatment for autoimmune disease in the light of several converging lines of evidence which have been reviewed elsewhere.22 ,23 Firstly, remission of their autoimmune disease is often seen in patients who undergo allogeneic, and more recently, autologous bone marrow transplant for haemopoietic or other malignancy. Secondly, evidence from disease susceptible strains of animals shows that autologous haemopoietic stem cells may cure the autoimmune disease and induce tolerance to the inciting agent. Lastly, the perception that immunosuppressive treatments using dosing regimens that remain sublethal are not effective in the treatment of some subjects with severe autoimmune disease suggests that the use of higher dose regimens requiring some form of bone marrow rescue may be superior.
Autologous haemopoietic stem cell transplantation as performed for haemopoietic or other malignancy carries a substantially lower transplant related mortality than allogeneic transplantation (5%v >15% respectively). Relative to the mortality associated with severe forms of autoimmune disease, this risk has been considered acceptable in view of the potential benefits of long term remission or the prevention of progressive tissue damage.22
Owing to the poor prognosis of SSc, a presumed significant autoimmune component, and the lack of available treatments, this disease was considered suitable for initial investigation of the tolerability and efficacy of autologous HSCT.
An international collaborative committee was established in 1995 under the auspices of the European Group for Blood and Marrow Transplantation (EBMT) and the European League Against Rheumatism (EULAR) and which later included other active groups in North America. Entry criteria and treatment protocols were established in phase I/II studies to assess feasibility, mortality, and preliminary response of such an approach.24 Most groups followed a core protocol written in accordance with these guidelines (HG Prentice), which allowed some flexibilty of local methodologies. The following report from a centralised database summarises the initial experience of this type of treatment for systemic sclerosis.
Patients and methods
STUDY DESIGN
The basic concept of these open, multicentre, phase I/II studies was described in a publication of the joint committee of EULAR and EBMT.24 Guidelines on patient selection generated as part of this consensus statement were provided to participating centres. Participants were requested to use a restricted number of protocols, though adherence to these protocols was not strictly controlled. The detailed treatment plans for individual patients followed local institutional protocols. All patients with SSc registered with the EBMT/EULAR International Stem Cell Project database up to August 1999 are included. Eighteen centres in eight countries contributed to this project. Although survival data are complete, in some cases not all clinical data were registered.
ELIGIBILITY
Patients with rapidly progressive early diffuse scleroderma (mostly <3 years' duration) who were assessed as having a high risk of further progression and mortality, in the absence of severe irreversible internal organ damage, were considered eligible. Patients with limited scleroderma were considered eligible if life threatening pulmonary fibrosis or pulmonary hypertension was present, and some of these had a disease duration of >3 years. Informed patient consent and approval by local ethical committees was a requisite for all protocols. Patients were informed that the expected early transplant related mortality was 3–5% and that the risk of death or severe disability from their disease was significantly higher.
TRANSPLANT TECHNIQUES
Peripheral blood stem cells were mobilised with one of three regimens using cyclophosphamide alone, cyclophosphamide (4 g/m2) followed by granulocyte colony stimulating factor (G-CSF), or G-CSF alone. In total, seven different conditioning regimens were used: 200 mg/kg cyclophosphamide; 120 mg/kg cyclophosphamide with anti-thymocyte globulin and total body irradiation (8 Gy); cyclophosphamide with anti-thymocyte globulin; cyclophosphamide with total lymphoid irradiation; cyclophosphamide with CAMPATH-1H (anti-CD52); busulphan and cyclophosphamide with anti-thymocyte globulin; BCNU (carmustine), fludarabine, and thiotepa.
OUTCOME MEASURES
Investigators were asked to provide information about mortality and disease progression. In addition, serial modified Rodnan skin scores and pulmonary (vital capacity (VC), carbon monoxide transfer factor (Tlco), and radiological data), cardiac (left ventricular ejection fraction (LVEF), electrocardiogram), and renal function (serum creatinine) test results were collected at three monthly intervals after transplant. When specific data were absent, outcome according to EBMT practice (complete remission, partial remission, no response, relapse, dead) was analysed.
STATISTICAL ANALYSIS
Statistical analysis of survival probability was actuarial using the Kaplan-Meier estimator. This means that patients with a short follow up have a reduced weighting compared with long term patients, important because new patients are continually being added to the database.
Results
PATIENTS
Over the period January 1996 to August 1999, 41 patients intended for autologous transplants for the treatment of SSc were reported to the EBMT/EULAR Registry. Four patients had stem cells mobilised but did not proceed to transplant. Thirty five patients underwent autologous peripheral blood stem cell transplant and two autologous bone marrow transplant. At the time of this report, all 41 patients had been followed up for at least three months after transplantation. Table 1gives details of patients.
Baseline characteristics of the cohort of patients with systemic sclerosis (SSc). The data are derived from the cohort of patients with SSc registered with EBMT for stem cell or bone marrow transplant. All patients with a minimum of three months' follow up data or who had died were included. In patients of disease duration >3 years, haemopoietic stem cell transplantation was performed owing to involvement of the vital organs Table 1(A) Basic demographic data and the duration of follow up
Thirty seven patients had diffuse cutaneous scleroderma. Twenty eight of these had some degree of pulmonary disease as judged by reduced Tlco and/or pulmonary infiltrate radiologically, seven were reported to have pulmonary hypertension, and five patients had renal involvement. Eighteen of 32 had anti-topoisomerase antibodies (Scl-70) and three had an anticentromere ANA pattern. Four patients had limited scleroderma with associated lung disease or pulmonary arterial hypertension. The median duration of disease at entry was 2.4 years (range 0.2–13). Tables 1(A), (B), and (C) show the baseline characteristics, measurements of organ function, and doctor's assessment of organ involvement for this group of patients.
PRIMING, CONDITIONING, AND PURGING
Table 2 shows the priming regimens (to induce mobilisation of stem cells from the bone marrow to the peripheral blood) used. One patient was not primed as an autologous bone marrow transplant was planned. Mobilisation was successful in all but one patient who subsequently received an autologous bone marrow transplant. Four patients died after mobilisation and before conditioning (see “Deaths”). The remainder received conditioning regimens as shown in table 2. No graft manipulation was performed on the bone marrow and three of the peripheral stem cell grafts. The other peripheral cell harvests were all positively selected for CD34+ cells before grafting. Some form of additional lymphocyte depletion was performed on a further seven grafts (CD52 alone (four), CD3 alone (one), CD2/3/19/20 (one), CD4/8 (one)). The purging protocol is unknown in one case. Engraftment was successful in all cases.
Treatment regimens employed
DISEASE OUTCOME
Responses
Of the 37 patients who received autologous bone marrow or HSCT transplants, the degree of disease response was reported by the supervising clinician in 27 of the surviving cases. Overall, positive clinical responses were recorded in 25 patients and no response was detected in four cases. A partial response to the priming regimen was noted in one patient such that transplantation was delayed. Disease progression was reported in seven patients, three of whom subsequently died, and six of whom had had initial positive responses. Another patient died of rapidly progressive cardiac disease before transplant could be performed, confirmed at necropsy. The median period from transplant to detection of progression was 67 days (range 49–555).
Skin scores
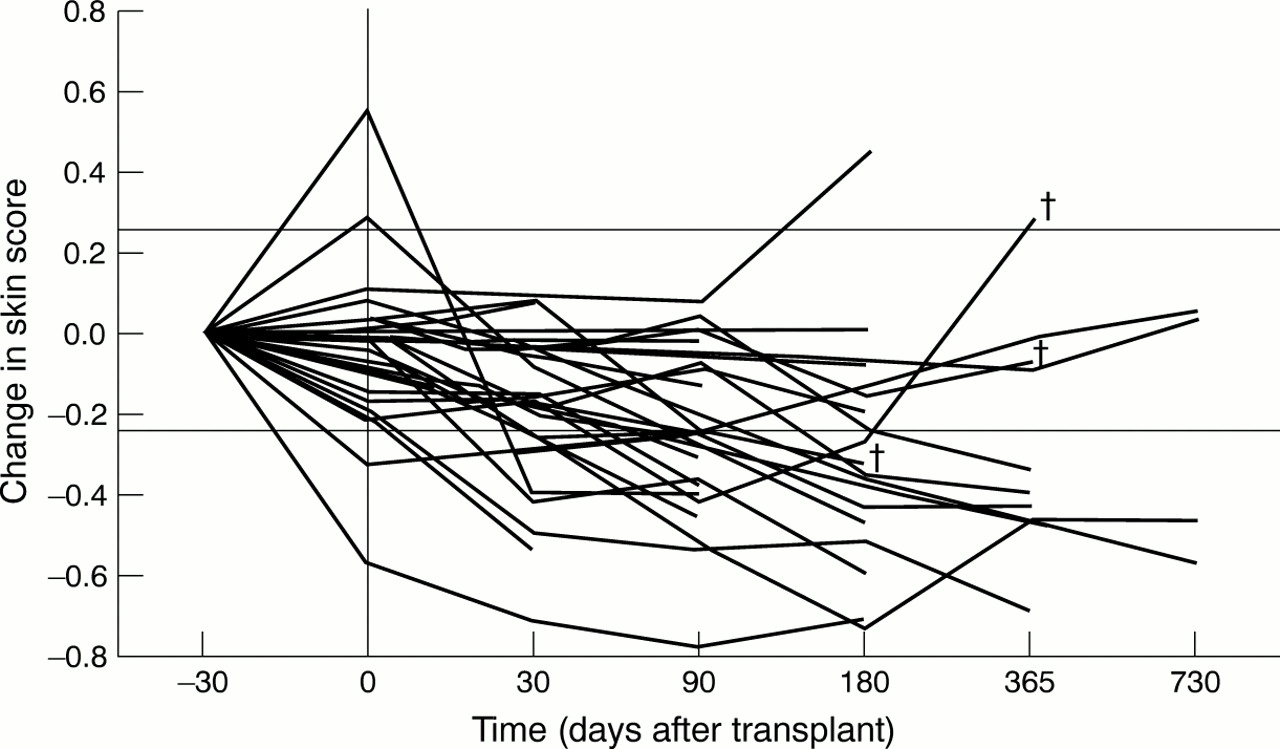
Of the 37 cases of diffuse scleroderma treated, serial skin score data were available in 29 cases (four patients died before the one month assessment and data were not registered in the other four). A decline in skin score was considered significant if it was greater than 25% of baseline or greater than 10% of the maximum skin score (three different modified Rodnan scoring systems were used). These changes are generally considered to be of clinical significance.25When this definition was used, 20/29 (69%) patients improved. Figure 1shows the change in skin score from baseline for each case. Two patients who initially improved subsequently died (one with progressive skin and pulmonary disease and the other with pulmonary hypertension). In two cases the skin score deteriorated. The mean skin scores at 30, 90, and 180 days were significantly less than those before mobilisation (p<0.005, paired Student's ttest).
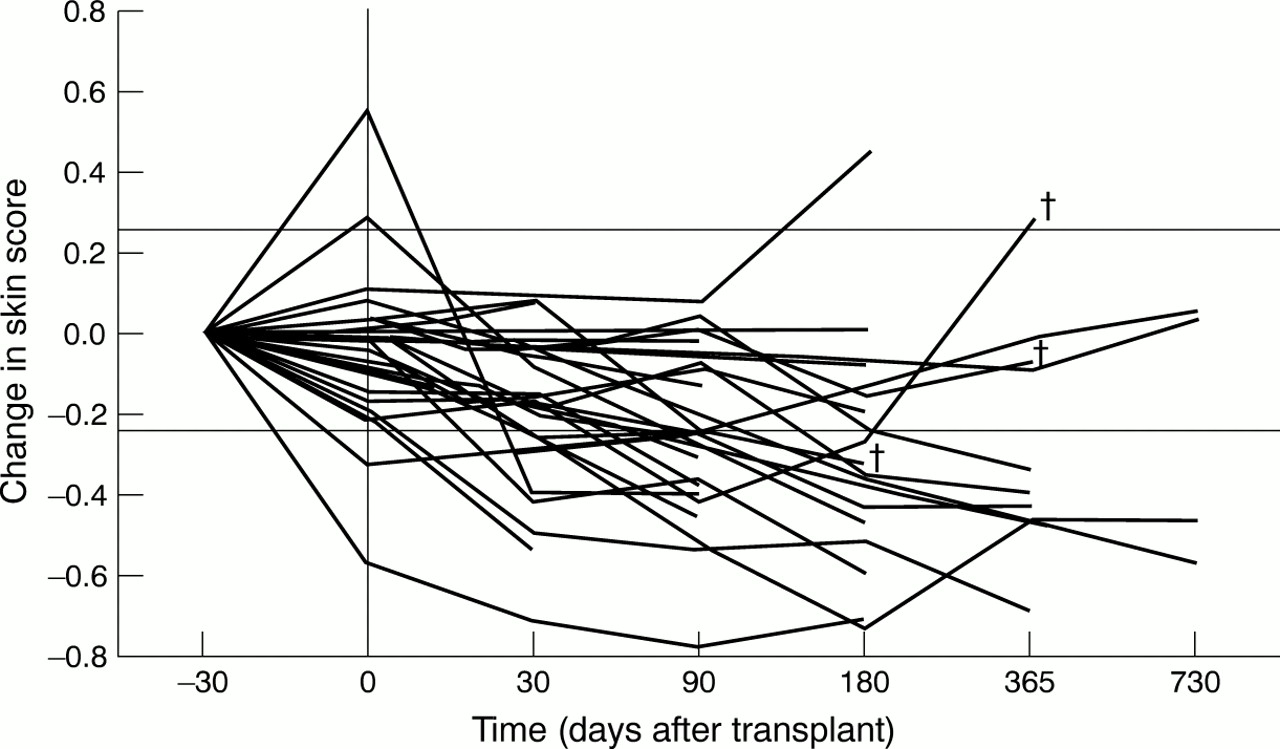
Change in skin score. The serial skin score data are presented for the 37 patients with diffuse scleroderma. The proportional change from baseline measurement was calculated for each patient at each available time point. Two horizontal lines are marked, which represent changes of 25%. The x axis is not drawn to scale. Data obtained before administration of the priming regimen are shown at –30 days (although the temporal relation to conditioning was variable). A vertical line is drawn to show the timing of conditioning treatment. † = patient death, but is only shown for deaths beyond 90 days.
Lung function
Measurements of VC and Tlco were available in 28 patients with early lung disease at baseline. At least three months' serial follow up data were available in 23 of these cases. A 15% change in VC or Tlco was considered to be clinically significant. When this criterion was used, the VC improved in 16% of patients, deteriorated in 24%, and remained stable in 68%. Some of this improvement might be due to improved chest wall skin thickness. The Tlco improved in 9% of patients, deteriorated in 39%, and remained stable in 52%. Of the nine patients who had no lung disease at baseline and for whom at least three month's follow up data were available, one had a significant fall in VC and Tlcoto below 70% predicted at three months. Despite a transient improvement in skin score, this patient subsequently died with progressive disease at one year.
Renal function
Five patients were reported to have scleroderma related renal impairment. One of the four patients for whom serial data were available deteriorated, with a 57% increase in serum creatinine at one year. The remainder were not significantly changed. Baseline creatinine clearance was available in 24 cases (mean (SD) 104.5 (29.5)). No significant correlation between baseline renal function and survival was found. Serial post-transplant data were available in 11 cases. The final serum creatinine measured showed an average increase of 10% over baseline (SD 22%).
Cardiac function
The left ventricular ejection fraction (LVEF) was measured before transplant in 26 cases. Mean (SD) LVEF was 61.15 (9.97)% for this group. No significant correlation was seen between baseline LVEF and survival. Of the nine patients in whom LVEF measurements were taken before and after transplant, the largest decline in LVEF after transplant was <10%. One patient with myositis but no clinical evidence of myocarditis had a significant drop in LVEF from 68% before to 35% three weeks after mobilisation treatment and subsequently died. Of four patients transplanted with baseline echo Doppler estimates of pulmonary pressure which were >30 mm Hg (but <50 mm Hg), all showed stable or improved measurements at three, six, or 12 months.
DEATHS
At the current census 11/41 (27%) patients with scleroderma enrolled with EBMT/EULAR died after the start of treatment. The median survival period of the cohort at census was 228 days (range 0–1663). Table 3 shows details of the deaths. Four patients died after priming but before transplant. One of these deaths was due to disease progression. Two were clearly related to the priming treatment (table3), and one sudden death appeared to be due to coincident ischaemic heart disease. All four of these patients received the priming regimen of cyclophosphamide 4 g/m2 plus G-CSF. Of the remaining seven deaths, four seemed to be directly related to the transplant. The two patients who died from pulmonary haemorrhage during the cyclophosphamide induced thrombocytopenic period had received apparently adequate platelet support, and this is an uncommon, but known, complication of HSCT. The other patient who died 40 days after transplant from cerebral haemorrhage had severe multisystem SSc from the beginning, including renal failure, and was not reported as thrombocytopenic. The haemorrhage was probably mulifactorial, including progressive disease and septicaemia, but was included in the procedure related deaths to avoid reporting bias. Of note were two deaths due to rapidly progressive interstitial pneumonitis occurring in a similar fashion—one three months after conditioning, which included total body irradiation. Figure 2 shows the Kaplan-Meier analysis of survival.
Deaths after the start of the procedure. The details relevant to the deaths of all patients registered with EBMT for haemopoietic stem cell transplantation in systemic sclerosis. The deaths are categorised by the relation to the procedure. In patients who died after the mobilisation regimen, but before conditioning, the survival time is listed as 0 days. Otherwise, the transplant date is taken as day 0

{kind=link}
{kind=link}
Cumulative probability of survival. A Kaplan-Meier plot of cumulative survival is shown. Survival data at census were analysed using SPSS software.
OUTCOME IN CHILDREN
Transplants were carried out in three children under the age of 18, all with scleroderma lung disease (two diffuse, one limited). All three are alive, and one has survived for two years with no deterioration in lung function measurements. The pulmonary outcome in the other two have yet to be assessed, but skin scores in the two patients with diffuse disease have significantly improved.26
Discussion
The data presented here were obtained from the EULAR/EBMT Working Party on Autoimmune Diseases database and derived from phase I/II pilot studies. The primary objectives were to assess toxicity and efficacy in a setting in which reporting biases of this new but hazardous treatment are minimised. In addition, an understanding of the effects of immunoablation on the progression of this profibrotic disease may advance hypotheses of disease pathogenesis, and, in particular, may illuminate the role of the immune system and/or other cells of haemopoietic origin in established disease.
The cohort of patients studied was gathered from many groups in several countries. Patient selection and therapeutic regimens were therefore varied, but all the treatment regimens were more severe than previously published. Although the cohort overall represented a group with severe disease and a poor prognosis, as judged by the skin scores, the proportion with lung disease, and carriage of the Scl-70 antibody, there was undoubtedly considerable heterogeneity in patient selection, resulting in a large variation in mortality rates between established transplant centres. Therapeutic regimens were identical in 73% of cases for priming (cyclophosphamide 4 g/m2 plus G-CSF) and 51% (cyclophosphamide 150–200 mg/kg) for conditioning regimens. It is assumed that profound immunosuppression was achieved in all those conditioned.
The effect of autologous stem cell transplantation on skin scores is striking. To our knowledge no other studies of therapeutic interventions in diffuse scleroderma have shown such an effect on the stabilisation of skin scores in patients with SSc with established disease. One randomised, double blind study comparing high and very low dose d-penicillamine in 134 patients with early (<18 months) scleroderma showed, in a modified intention to treat analysis, that numbers of responders (>25% improved skin score) were 44% and 38% in the high and low dose groups, respectively. In those completing the 24 months of drug treatment, these figures were 70% and 53%, respectively.18 The authors concluded that skin thickness in patients with early SSc may improve spontaneously as the disease moves from an oedematous/inflammatory phase through collagen overproduction and eventually atrophy. The patients in our report were at a later stage of their disease (median disease duration 2.4 years), with a progressive course, and it is unlikely that the mostly sustained improved skin score represented a purely anti-inflammatory effect or a natural resolution.
It should be borne in mind that skin scoring is susceptible to observer bias and in an unblinded study of such a major intervention, this may be significant. However, despite this limitation, similar patterns were recorded across many experienced centres.
No data on pretreatment rates of change of skin score were available, but after the priming treatment 13% of patients showed some evidence of skin progression. The maximum skin score increase in the three months after transplantation was 6.9%. Over this period 91% of patients were unchanged or improved. At six months 94% of cases recorded no deterioration. Two patients escaped control between three and six months after transplant and continued to progress. Within the limited data available, there is no indication as to why these and other patients relapsed, including immune reconstitution (“memory” CD45RO and helper CD4 cell numbers), pretransplant, state or type of regimen. The planned prospective, randomised trial comparing monthly cyclophosphamide with HSCT will attempt to deal with this issue. For those patients with one year follow up data, 90% had not deteriorated. However, at two years after transplant, two of four patients had developed progressive skin disease, one of whom died of progressive disease. Thus the durability of the suppression of skin activity remains an open question because the duration of follow up of this cohort is too short for a meaningful assessment of the frequency of reactivation of skin disease. Also, it is not clear from this study if the benefit is due to some specific feature of HSCT or cyclophosphamide alone.
As mentioned above, data on lymphocyte numbers and phenotype after transplant (not shown) do not support the concept that relapse of disease occurs when the immune system returns to “normal”. However, some patients followed up for >4 years and remaining in remission have sustained low CD4 T cell numbers. One of these patients had two episodes of pneumonia (no organism identified) and required prophylaxis with co-trimoxazole to stabilise. More immune reconstitution data are required. Although lymphocyte recovery may be critical, neutrophil and macrophage/monocyte depletion may also contribute to the effect on the skin score. The recovery of these groups, however, is more rapid. Another feature of immunoablative chemotherapy is the reported decline in circulating levels of basic fibroblast growth factor,27which may contribute to the improvement in skin fibrosis seen.
The pulmonary fibrosis in SSc is responsible for about half of the mortality of the condition. Immunoablative chemotherapy might be predicted to have an effect on this condition as there is good evidence of immunological involvement in its pathogenesis28 ,29 and it is thought to respond to immunosuppressive regimens. Although there was overall no statistically significant improvement in pulmonary function in this cohort, stabilisation, improvement, and deterioration were all seen, with a trend towards stabilisation in a group at risk for progression. It is as yet impossible to tell whether this is related to the regimen, a patient selection effect, or some combination of these. Only careful systematic attention to SSc regimens (especially prospective, randomised large number trials) can lead to rational decisions on the effect of HSCT on pulmonary function. Similarly, the numbers of patients with pulmonary hypertension remain too small to interpret follow up data.
The transplant related mortality was higher than expected for the procedure. Estimates based on the use of autologous HSCT for haematological malignancy of 3–5% clearly do not take account of the multiple organ, vascular, and immunological abnormalities found in SSc. Experience with the procedure in other autoimmune diseases30 may suggest a slightly higher transplant related mortality than expected but not to the level of that found in SSc The transplant related mortality must be viewed in the light of a disease with significant five year mortality of the order of 50% or greater in certain high risk patient subsets,17approximately equal to that of chronic myeloid leukaemia in the chronic phase. The patients selected for this study of an extreme experimental therapeutic regimen had severe disease. The deaths clearly related to disease accounted for 36% of the deaths in this study at this census. Although the total mortality of this cohort is currently 27%, and the follow up period remains short, the procedure related mortality at 17% is unlikely to rise further. Indeed, when the proposed exclusion criteria derived from this experience are used (see below), three of the patients who had procedure related deaths and one of the patients with progressive disease would have not have received a transplant. This would have resulted in a procedure related mortality of 10.5% and overall mortality of 19%. Also, since submitting this manuscript, a further 24 patients with SSc who have received a transplant have been registered (with only one fatality), resulting in a total procedure related mortality of 12% (data from the EBMT/EULAR database). This probably reflects a learning process about patient selection. When the whole 65 patient cohort is considered, the recalculated procedure related mortality with patient exclusion according to the current guidelines is 5/65 (8%).
Three critical questions arise: (a) If this cohort had received conventional treatment, what would the mortality rate have been? (Does HSCT improve survival?); (b) Will future follow up show a reduced mortality rate from disease progression? (Can an increased risk of early mortality be justified by improvements in the long term?); (c) Can the procedure itself be made any safer?
Answers to the first two questions can only be obtained from a randomised controlled trial. The safety of the procedure might be improved by careful case selection and possible modification of the regimen. A consensus meeting of the investigators highlighted the exclusion of patients with pulmonary artery pressure measurements >50 mm Hg, significant myocardial disease (ventricular ejection fraction of <50% predicted and/or uncontrolled arrhythmias), established lung disease with Tlco <45% of predicted, and gastrointestinal involvement requiring total parenteral nutrition. Ideal candidates would be those with diffuse skin disease of <4 years' duration and involvement of at least one organ system (pulmonary, cardiac, renal). Progression despite conventional treatment and the development of new organ disease would add to the indication. The therapeutic regimen should be modified to take account of the four deaths after mobilisation, all of which occurred in the group receiving 4 g/m2 cyclophosphamide. Reduction of this dose may reduce the risk, though those patients unable to tolerate this treatment are unlikely to tolerate the conditioning regimen. Some of the risk may be due to the cardiotoxicity of the drug and careful screening for evidence of SSc related myocarditis or cardiomyopathy. The avoidance of cyclophosphamide in the conditioning regimen in those patients with cardiac disease might be advisable. The second recommended change would be the avoidance of total body irradiation without the use of lung shielding. The two cases of rapidly progressive interstitial pneumonitis after transplant might have been related to radiation exposure or some combination of cyclophosphamide related toxicity and total body irradiation in patients with fibrotic lung disease.
These phase I/II studies have demonstrated an impact on skin progression, a trend towards stabilisation of lung disease, but with significant toxicity of treatment. Given this, and the considerable problems of comparison with other cohorts of patients, it is clear that only a randomised controlled study of autologous HSCT in SSc will ultimately provide information critical to decisions about the costs and benefits of this approach to treatment.
Acknowledgments
We thank Chiara Bocelli-Tyndall, project manager, for gathering and collating the data.
Contributing centres
Royal Free Hospital, London, UK (MB, HGP, CB), Kantonsspital Basel, Switzerland (JRP, AG, AT), Fred Hutchinson Cancer Research Centre, Seattle, WA, USA (DF, PMS, KS), Albert Ludwig Universität, Freiburg, Germany (JF, HHP, CB), Akademish Ziekenhuis, Leiden, The Netherlands (JvL, FCB, WEF), Hôpital St. Louis, Paris, France (DF, EG), Ospedale San Matteo, Pavia, Italy (AM, FL), City of Hope Medical Centre, Duarte, CA, USA (AK), University Hospital Nijmegen, The Netherlands (FvdH, LvdP, AVNS), University of Birmingham, UK (PAB), University of Leeds, UK (PE), Charité Hospital, Berlin, Germany (RA, FH), Hospital Carlos Haya, Malaga, Spain (MJP), Hospital Universitario Virgen del Rocio, Seville, Spain (IE), Hospital Infantil La Paz, Madrid, Spain (AM), Universitätsklinikum Tübingen, Germany (IK), Medizinische Hochschule, Hannover, Germany (BH), Ospedale San Martino, Genova, Italy (AM).
References
Footnotes
Details of the contributing centres are given in the .
The work was partly supported by an educational grant from Amgen for the mobilisation data, F Hoffman-La Roche for travel support of the writing committee, and Swiss National Research Fund Grant No 31-45938.95 for establishing the database.