Article Text

Abstract
Objective: Evaluate long-term safety and efficacy of etanercept treatment in patients with ankylosing spondylitis (AS).
Methods: Patients with AS who previously participated in a randomised controlled trial (RCT) of etanercept were eligible to enrol in a 168-week open-label extension (OLE). Safety end points included rates of adverse events (AE), serious adverse events (SAE), infections, serious infections and death. Efficacy end points included Assessment in Ankylosing Spondylitis (ASAS20) response, ASAS 5/6 response and partial remission rates.
Results: A total of 257 of 277 patients (92%) enrolled in the OLE. After up to 192 weeks of treatment with etanercept, the most common AEs were injection site reactions, headaches and diarrhoea. The exposure-adjusted rate of SAEs was 0.08 per patient-year. The rate of infections was 1.1 per patient-year, and the rate for serious infections was 0.02 per patient-year. No deaths were reported. Of patients who received etanercept in both the RCT and OLE and were still in the trial, 71% were ASAS20 responders at week 96, and 81% were responders at week 192. ASAS 5/6 response rates were 61% at week 96 and 60% at week 144, and partial remission response rates were 41% at week 96 and 44% at week 192. Placebo patients who switched to etanercept in the OLE showed similar patterns of efficacy maintenance.
Conclusions: Etanercept was well tolerated for up to 192 weeks in patients with AS, with no unexpected AEs or SAEs observed. No deaths were reported. Improvements in the signs and symptoms of AS were maintained for up to 192 weeks.
Statistics from Altmetric.com
Ankylosing spondylitis (AS) is the most prevalent and severe of the spondyloarthropathies, which share common characteristics of sacroiliitis, enthesitis, uveitis, psoriasis and inflammatory bowel disease (IBD). AS primarily affects the spine and is characterised by enthesitis, ankylosis of the axial skeleton and occasionally peripheral arthritis. Disease progression can lead to fusion of the spine, loss of mobility and loss of function. In turn, these restrictions can lead to decreased daily activity and severely reduced health-related quality of life.1 Cornerstone treatments of AS include physiotherapy, exercise and non-steroidal anti-inflammatory drugs (NSAIDs), which are used to manage the symptoms of AS. However, there is little evidence that these therapies produce long-term disease benefit.1–5
Tumour necrosis factor (TNF) plays an important part in the inflammatory pathology of AS, with AS patients demonstrating increased levels of TNF in serum and joint synovium.6 7 Controlled studies have demonstrated that TNF blocking agents (etanercept, adalimumab and infliximab) safely and effectively reduces the signs and symptoms of AS8–17 and significantly improve health-related quality of life in AS patients.1 Specifically, results from a random controlled trial (RCT) have demonstrated that etanercept (Enbrel), a fully human soluble TNF receptor antagonist was well tolerated by patients with AS and was highly effective at reducing the signs and symptoms of the disease for up to 24 weeks.12 Data from an open-label extension (OLE) have demonstrated continued safety and efficacy of etanercept for up to 2 years in patients with AS.16 In the current report, the long-term safety and efficacy profiles of etanercept are reported for patients with AS receiving etanercept therapy for up to 192 weeks.
PATIENTS AND METHODS
Patients and study design
The randomised, double-blinded, placebo-controlled trial12 and the 96-week OLE14 have previously been described. In brief, the RCT enrolled a total of 277 AS patients (placebo, n = 139; etanercept, n = 138) for 24 weeks of treatment. Eligible patients were then given the option to enrol in an OLE, which was extended for a total of 168 weeks. Therefore, patients who received etanercept treatment in the initial RCT potentially received up to 192 weeks of continuous treatment with etanercept, and those who received placebo in the initial trial could receive up to 168 weeks of continuous treatment with etanercept.
This study was conducted in accordance with the Declaration of Helsinki, was approved by the IRB (US) and IEC (Canada and Europe) and informed consent was obtained from all patients.
Stable doses of NSAIDs and corticosteroids (⩽10 mg/day) were required 2 weeks prior to enrolment. Stable dosing of methotrexate, hydroxychloroquine or sulfasalazine were required if concomitant use was deemed necessary. Use of other disease-modifying anti-rheumatic drugs (DMARDs) needed to be discontinued 4 weeks prior to enrolment. Previous treatment with any anti-TNF agent other than etanercept was not permitted.
Study medication
Etanercept was originally administered subcutaneously as two separate doses of 25 mg given twice per week for a total of 50 mg of medication weekly. After week 72 of the OLE study, 50 mg of etanercept was administered as two 25 mg subcutaneous injections given once weekly.
Procedures and end points
Safety data collected included adverse events (AEs), serious adverse events (SAEs), infections, serious infections, and death.
In this completer’s analysis, clinical efficacy was determined using criteria defined by the Assessment in Ankylosing Spondylitis (ASAS) International Working Group for response (ASAS20 response) and partial remission.18 ASAS40 response also was evaluated. An ASAS40 response requires a 40% and 20 unit improvement in at least three of the four criteria included in the ASAS20 response (PtGA, pain, function, inflammation) with no deterioration in the remaining criterion.
The “ASAS 5 out of 6” (ASAS 5/6) response was analysed. The ASAS 5/6 includes the domains of the ASAS20, but also includes two additional domains: spinal mobility and acute phase reactant levels. Therefore, ASAS 5/6 responses requires a ⩾20% improvement in five of six domains including: PtGA, pain, function, inflammation, C-reactive protein and spinal mobility (chest expansion).
Each individual component of the ASAS20 response (PtGA, pain, function and inflammation), total Bath Ankylosing Spondylitis Functional Index (BASFI) and total Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) were analysed independently as well. Spinal mobility was assessed by the modified Schober test, chest expansion, and by the occiput-to-wall distance.
ASAS20 response, ASAS40 response and partial remission were also calculated for the subjects who had at least one post-dose assessment, using LOCF for missing data imputation.
Statistics
The proportions of patients meeting the end points were reported over time for each treatment group: patients receiving continuous etanercept (ETN/ETN) and those who received placebo for 24 weeks of the RCT and etanercept in the OLE (PBO/ETN). Statistical inferences regarding efficacy were not made between the two groups. All baseline values reported were determined for the initial RCT. Efficacy values reported represent a summary of data from all patients who had data available at that time point. Data for all of the individual components of the indices were collected at 12- or 24-week intervals for the entire 168-week OLE, except for chest expansion, which was not recorded for all patients at the last time points, limiting ASAS 5/6 assessment to 144 weeks.
RESULTS
Patient characteristics
A total of 277 patients enrolled in the RCT; 139 patients received placebo and 138 received etanercept. A total of 257 patients enrolled in the OLE; 129 from the placebo group and 128 from the etanercept group. Demographic data are shown in Table 1.
All 257 patients in the OLE study received 50 mg of etanercept weekly for 168 weeks or until early termination. Each of the 257 patients received at least one dose of etanercept. The total number of patient-years of etanercept treatment was 650 combining the patient exposure time from the RCT and the OLE or 587 for the OLE alone.
Figure 1 shows the study design and patient discontinuation. A total of 126 patients (49%) completed the 168-week OLE. The mean baseline characteristics of the patients who completed the OLE and of all the patients who enrolled in the OLE were similar (table 1). A total of 34 patients (13.2%) did not re-enrol in the OLE when amendments were instituted to lengthen the study duration. The other most common reasons for study withdrawal were patient refusal (10.1%) and AEs (8.2%, including infections and injection site reactions (ISRs)).

Safety
Table 2 summarises the total number of AEs, SAEs, infections and serious infections and compares the incidence of these events for patients receiving placebo in the RCT to all patients receiving at least one dose of etanercept in the OLE. ISRs were the most common AE reported by patients receiving etanercept (22.2%), followed by headaches (20.2%), diarrhoea (17.5%), injury accident (16%) and pain (14.4%). A total of 14 patients (5.4%) withdrew from the OLE because of AEs excluding infectious episodes and ISRs; only rash led to the withdrawal of more than one patient (n = 2).
AEs of interest included events relating to the eye (uveitis). A total of 44 of 139 patients (32%) receiving placebo in the RCT had a history of uveitis prior to enrolment. Patients receiving placebo in RCT reported 13 flares and no new incidence of uveitis for an exposure adjusted rate of flares (including new incidents) of 0.22 per patient-year. Of all patients who received at least one dose of etanercept in the RCT or OLE, 80 of 267 patients (30%) had a history of uveitis prior to the start of the RCT. Of the patients receiving etanercept in the RCT or the OLE, 62 flares and 10 new incidence of uveitis were reported for an exposure adjusted rate of flares (including new incidents) of 0.11 per patient-year. The exposure adjusted rate of new episodes of uveitis was 0.02 per patient-year.
Other AEs of interest were events relating to IBD. In the RCT, a total of 13 of 139 patients (9%) receiving placebo had a history of IBD prior to enrolment. No flares were reported in these 13 patients over 24 weeks. One new episode of IBD occurred in a patient receiving placebo during the RCT for an exposure-adjusted rate of new cases of IBD of 0.019 per patient-year. Of all patients who received at least one dose of etanercept in the RCT or OLE, 23 of 267 patients (9%) had a prior history of IBD before enrolling in the RCT. A total of 11 flares were reported in eight of the 23 patients (34.8%) who had prior IBD or developed IBD over the course of the RCT and OLE for an exposure adjusted rate of flares of 0.21 per patient-year. New incidents of IBD occurred in two of 244 patients (0.8%) who received at least one dose of etanercept and had no history of IBD prior to the RCT, with one of these new occurrences occurring during the RCT and the other occurring after 2.3 years of etanercept exposure. Of note, the patient with a new diagnosis of IBD in the OLE had a history consistent with IBD with abdominal pain and diarrhoea prior to receiving etanercept but without specific diagnosis. The exposure-adjusted rate of new cases of IBD in patients receiving etanercept was 0.003 per patient-year.
Rates of SAEs were 0.08 per patient-year in patients receiving etanercept and 0.07 per patient-year in patients receiving placebo. The most common SAE reported by etanercept patients was depression (n = 3). Back pain, malnutrition and fatty liver were each reported twice in patients receiving etanercept; no other SAE was reported more than once. No SAEs were reported relating to lupus, demyelinating disorders or lymphomas.
The exposure adjusted rate of infections was 1.0 per patient-year for patients receiving placebo and 1.1 per patient-year for patients receiving etanercept. A total of four patients (1.6%) withdrew because of infections. The most common infections reported were upper respiratory (45%), sinusitis (16%), flu syndrome (15%) and bronchitis (11%). The exposure-adjusted rate of serious infections was 0.02 per patient-year for each group. A tuberculosis event, confirmed via biopsy, was reported in one patient (0.4%) following 2.8 years of etanercept exposure; etanercept was discontinued. The patient was born in a country where tuberculosis is common and about 3 years before the event the patient had a negative purified protein derivative test as well as a negative chest image.
Efficacy
Proportion of subjects achieving ASAS20 response and ASAS40 responses was calculated over time (fig 2). ETN/ETN patients demonstrated a durable response over 192 weeks of etanercept treatment. A total of 48% of patients achieved an ASAS20 response and 30% achieved an ASAS40 response by week 2. After 192 weeks of etanercept exposure, the ASAS20 response was 81% and the ASAS40 response was 69%. ETN/ETN patients (N = 124) analysed using LOCF showed response rates of 67% for ASAS20 response and 49% for ASAS40 response when carried out to 192 weeks.
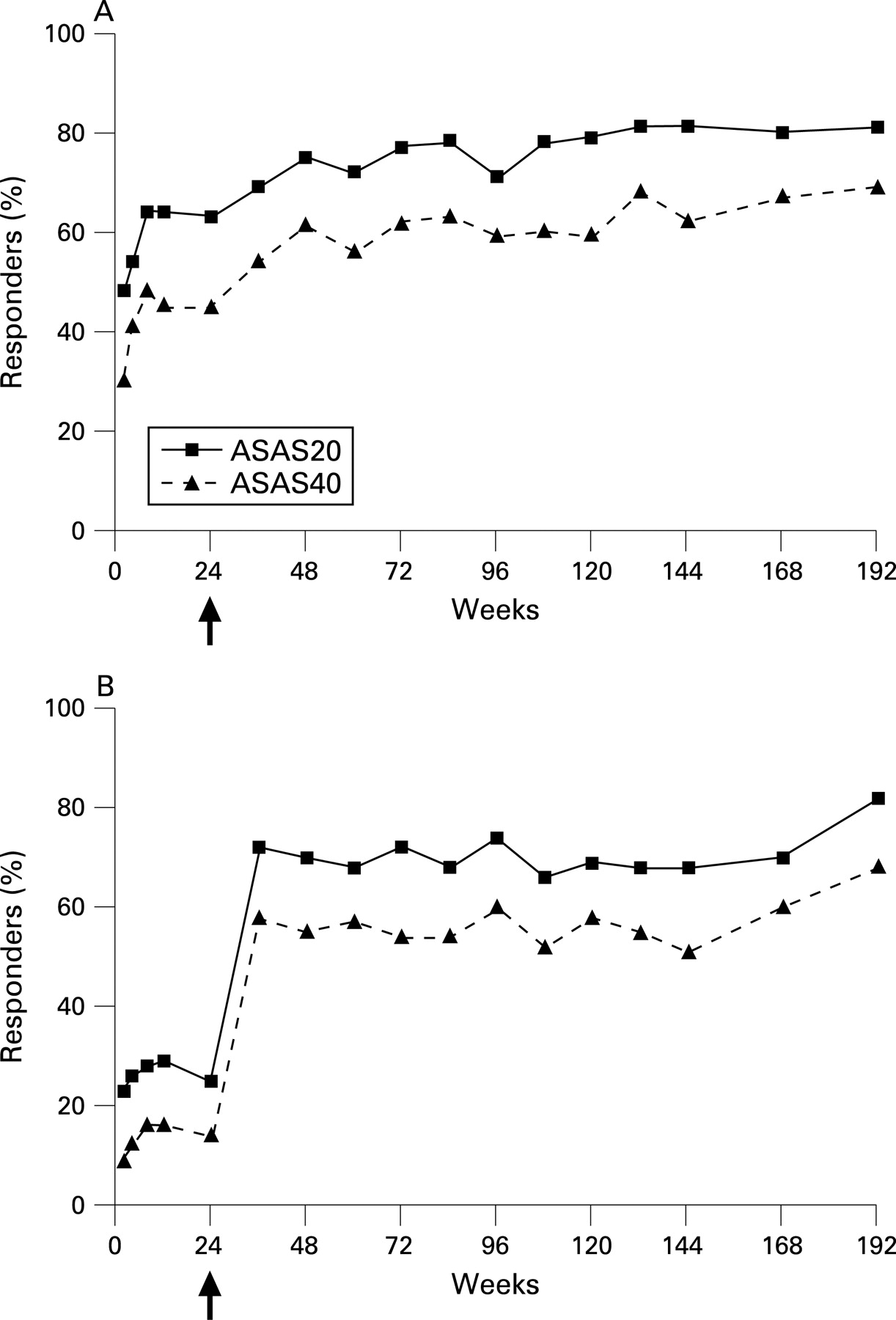
PBO/ETN patients demonstrated a sustained response once etanercept treatment began (fig 2). After 168 weeks of continuous etanercept exposure, the ASAS20 response was 82% and the ASAS40 response rate 68% using a completers’ analysis. Response rates of the PBO/ETN patients (N = 127) using LOCF were 61% for ASAS20 response and 50% for ASAS40 response.
The early pattern of the ASAS 5/6 response rate was similar to those of the other indices (fig 3). ETN/ETN patients had durable ASAS 5/6 response rates: 49% were responders by week 8 and 60% were responders at week 144. PBO/ETN patients quickly reached response rates close to those of the ETN/ETN patients following the switch to etanercept. A total of 48% were ASAS 5/6 responders at week 48 (week 24 of active treatment) and 40% at week 144 (week 120 of active treatment).
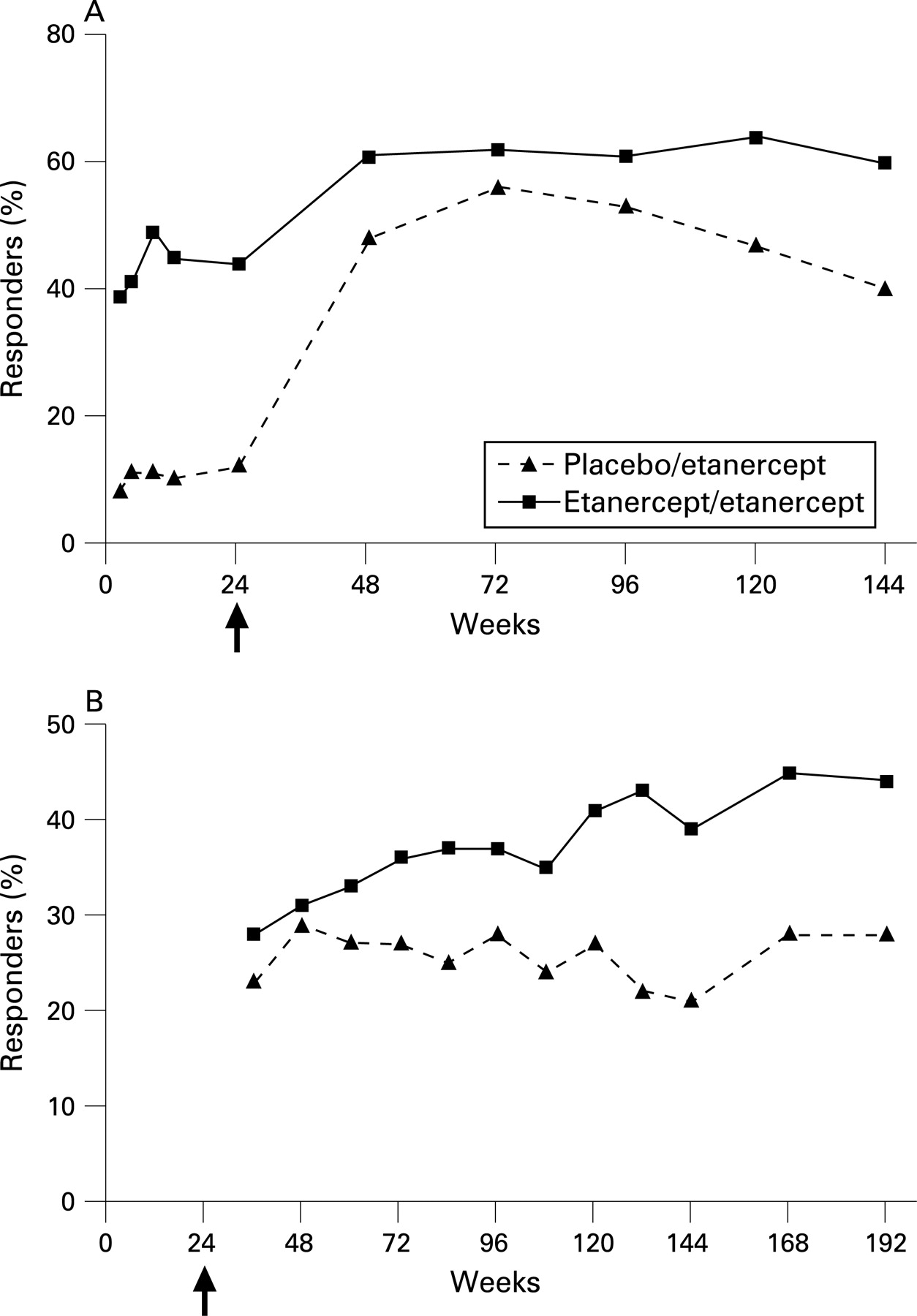
Partial remission rates also were maintained throughout the length of the OLE (fig 3). Partial remission was achieved by 44% of the ETN/ETN patients and 28% of the PBO/ETN patients at week 192. Partial remission rates using LOCF for the ETN/ETN patients and PBO/ETN patients were 27% and 21%, respectively.
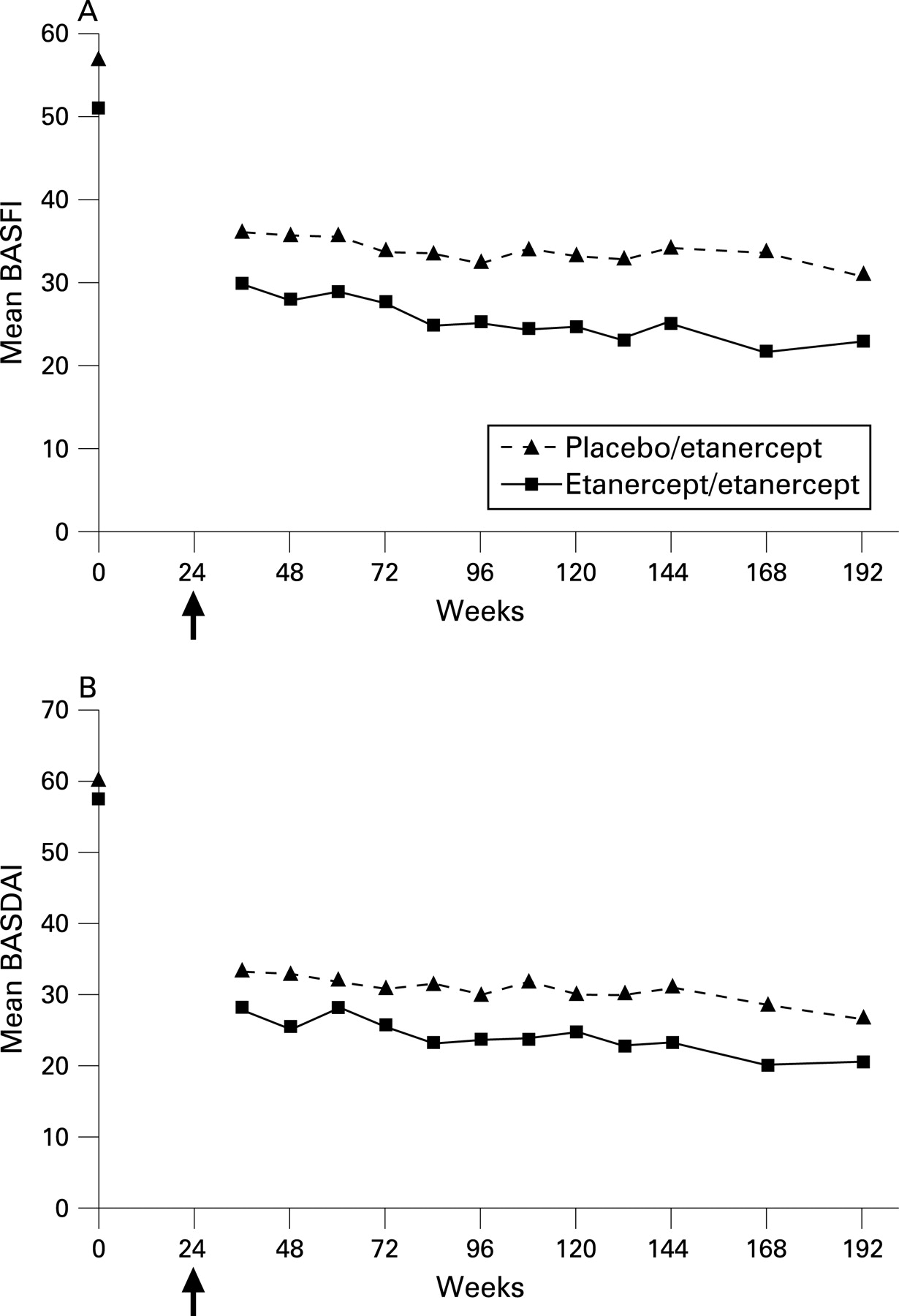
Results of the analyses of the individual components of the multiple composite scores are listed in table 3, and fig 4 shows mean BASFI and mean BASDAI scores over time. In ETN/ETN patients, all measurements were improved from baseline at the first follow-up of the OLE and maintained through the end of the OLE trial. All baseline values for the PBO/ETN patients were improved by the first follow-up of the OLE except one spinal mobility measure, which remained unchanged. After 168 weeks of etanercept exposure, only the modified Schober test score and the occiput-to wall measure were not improved over baseline values for the PBO/ETN patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
The current results from this 192-week OLE provide evidence that the generally favourable safety and efficacy profiles of etanercept in AS are maintained long term. No unexpected AEs, SAEs, serious infections, or deaths were reported. In addition, efficacy as measured by individual measures of disease activity as well as composite indices showed sustained improvements with etanercept treatment.
In this study, 126 patients (49%) completed the 192-week OLE. This is lower than expected and is most likely due to the 34 patients (13.2%) who discontinued the OLE by not re-enrolling when amendments to prolong the OLE were added. In total, only 7.8% of patients discontinued because of a lack of efficacy and only 8.2% of patients discontinued because of an AE, infection or ISR. In addition, while 45 of 89 patients (51%) that had baseline use of the allowed concomitant DMARDs completed the OLE, a percentage similar to the percentage of all patients completing the OLE, only 22 of 45 (49%) of these patients were still receiving these DMARDs by weeks 145–168 of the OLE.
As previously reported, the most frequent AE was ISRs.16 ISRs were generally mild or moderate and decreased in frequency over time; only four additional patients (2%) reported ISRs during the last 96 weeks of the OLE. Uveitis flares occurred at a rate of 0.11 per patient-year of etanercept exposure, lower than the rate of flares reported by placebo patients in the RCT (0.22 per patient-year). Previous reports indicated that AS patients receiving etanercept therapy reported significantly fewer flares of anterior uveitis than patients receiving placebo (7.9 vs 15.6 per 100 patient-year).19 While this analysis does not have the power to address this issue directly, the rates of flares reported in this analysis for patients receiving etanercept therapy are similarly low. AS patients in this analysis showed no unexpected events related to IBD, as the exposure adjusted rate of new reported incidents of IBD in patients receiving placebo in the RCT (0.019) was greater than the exposure adjusted rate of new reported incidents of IBD in patients receiving etanercept up to 192 weeks throughout the RCT and OLE (0.003).
During this nearly 4-year-long study, the average rate of SAEs was 0.08 per patient-year. This rate is similar to the rate of 0.07 per patient-year seen in patients receiving placebo in the RCT. This value is also less than the rate of 0.15 per patient-year reported in patients with rheumatoid arthritis over a 7-year period of etanercept treatment20 or the rate of 0.13 per patient-year reported in patients with juvenile rheumatoid arthritis over a 4-year period of etanercept treatment.21 Differences in underlying diseases, age of the patients, comorbidities and concomitant therapy may explain the difference in SAEs rates.
As with the 96-week OLE, the most common type of infection reported was upper respiratory infections. Infections led to the withdrawal of four (1.6%) patients from the study, two during the first 96 weeks of the OLE and two after. A total of six AS patients (2.3%) in this analysis experienced a serious infection. The rate of 0.02 serious infections per patient-year for patients receiving etanercept in the OLE was similar to the rate seen in placebo patients of the RCT (0.02 per patient-year). Active infections should be monitored and appropriately treated if found. The overall safety profile of infections with long-term etanercept treatment of patients with AS is comparable with the long-term safety profiles of infections with etanercept use in patients with rheumatoid arthritis and juvenile rheumatoid arthritis. In addition, previous reports suggest that etanercept has a longer time-to-onset of tuberculosis than the anti-TNF antibodies and the onset of the single case of tuberculosis reported after 2.8 years of exposure reflects these findings.22–24
In this analysis, durable efficacy of etanercept was demonstrated by response rates being sustained over time for all indices used to measure disease activity, regardless of whether patients received placebo or etanercept in the RCT. For example, an ASAS20 response was achieved in >80% of all patients remaining in the study after 192 weeks. Assessments of disease activity with more stringent criteria, such as partial remission, and ASAS 5/6 response, also demonstrated durable efficacy. The proportion or patients achieving these more stringent measures of disease activity appeared to be greater for those who had received etanercept during the RCT. This may be because of earlier exposure to etanercept treatment, as it has previously been shown that AS patients with shorter disease duration respond significantly better to treatment with TNF blockers;25 though this study does not have the power to test this hypothesis.
Analysis with LOCF to impute missing values carried out to 192 weeks also yielded durable responses as 61–67% of all patients had an ASAS20 response. LOCF response rates were lower than response rates of the completers’ analysis; however, while each statistical approach has its own limitations, both measures show a durable response to etanercept with long-term use.
Finally, all patients demonstrated significant improvements in BASDAI, BASFI, PtGA, total back pain, morning stiffness, C-reactive protein, swollen joint count, and tender joint count scores after etanercept treatment, and these improvements were shown to be durable for up to 192 weeks. While all measures of spinal mobility showed improvements in patients who had initially received etanercept treatment in the RCT, in patients who initially received 24 weeks of placebo prior to etanercept therapy, only chest expansion scores were significantly improved. This difference between the two treatment groups may again reflect the earlier exposure to etanercept by those patients who received etanercept in the RCT or the longer exposure time the patients who received etanercept during the RCT had to etanercept.
In conclusion, these current data demonstrate the durable safety profile of long-term etanercept treatment in patients with no unexpected AEs being reported in up to 192 weeks of therapy. In addition, the data demonstrate that long-term treatment with etanercept results in sustainable clinical improvements in the signs and symptoms of AS for up to 192 weeks.
Acknowledgments
We thank Marc D Kubasak, PhD, for assistance with preparation of the manuscript.
REFERENCES
Footnotes
Funding: This study was funded by Immunex Corporation, a wholly owned subsidiary of Amgen Inc., and by Wyeth Pharmaceuticals.
Competing interests: DMvdH, JB and MD have been reimbursed by Amgen/Wyeth, the manufacturer of etanercept, for attending conferences. RDI, RMF, JCD, DMvdH, DOC, JB and MD have received fees as consultants or speakers; RMF, JCD, DMvdH, DOC, JB, AJK and MD have received research grants from Amgen/Wyeth; LN, S-L L and WHT are employees of Amgen Inc.