Article Text

Abstract
Rheumatoid arthritis (RA) is one of the most appropriate conditions for the application of personalised medicine as a high degree of heterogeneity has been recognised, which remains to be explained. Such heterogeneity is also reflected in the large number of treatment targets and options. A growing number of biologics as well as small molecules are already in use and there are promising new drugs in development. In order to make the best use of treatment options, both targeted and non-targeted biomarkers have to be identified and validated. To this aim, new rules are needed for the interaction between academia and industry under regulatory control. Setting up multi-centre biosample collections with clear definition of access, organising early, possibly non-committing discussions with regulatory authorities, and defining a clear route for the validation, qualification and registration of the biomarker–drug combination are some of the more critical areas where effective collaboration between the drug industry, academia and regulators is needed.
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a complex disease with a high degree of clinical heterogeneity, as assessed by genetics, environmental risk factors, autoantibody patterns, cytokine expression, clinical course and response to therapy. Because of the chronic nature of the disease, the societal cost is enormous and continues to rise. Over the last 10 years, better understanding of the pathogenesis of RA has led to treatments that are more effective. However, limitations include a heterogeneous response to treatment, potentially serious adverse events and lack of lasting remission.
Despite the availability of highly effective treatment options, inter-individual variation in disease onset, progression and response to therapy makes treatment of RA difficult. Timing of effective treatment is essential for a good outcome and for the prevention and retardation of joint damage. Maintaining a clinical balance between applying timely and effective treatment and avoiding ineffective, costly and potentially aggressive treatment is at present one of the main challenges in RA management. A tailored approach to treatment can be envisioned, based, for example, on combinations of biologics or sequential therapies guided by biomarkers.1 Unfortunately, the optimal tools for diagnosis, prognosis, treatment selection and efficacy measurement are not yet at hand.
The Group for the Respect of Ethics and Excellence in Science (GREES) consists of members from academia, industry and regulatory bodies. It holds meetings aimed at developing a strategy and defining the processes required to generate tools and prediction models for decision making in clinical practice, as recently for RA trials.2 The present GREES report is focused on biomarkers and personalised medicine designed to achieve this goal in RA. The objective was not to list all biomarkers but rather to express a consensus view from academia, industry and regulatory bodies.
Current use of biomarkers in RA
During the course of the disease, there are three major time points when crucial decisions are required. First, RA patients need to be diagnosed very early in the disease process, possibly before diagnostic criteria are fulfilled or maybe even before clinical symptoms are apparent (asymptomatic or preclinical stage). Markers for prognosis are especially needed at this stage. Second, in the case of early arthritis, markers of disease activity and severity are needed. Finally, screening tests for prediction of response to therapy and progression of the disease can be used to establish rules to increase treatment success and reduce safety concerns.
The biomarkers currently used for the diagnosis of RA are mostly clinical. The first biomarker used for RA was the presence of rheumatoid factors. However, this marker lacks specificity and has been supplemented by the detection of antibodies against cyclic citrullinated peptides, now referred to as anticitrullinated protein antibodies (ACPA). The first genetic marker found for RA is in the HLA-DR region. HLA-DR molecules associated with RA are HLA-DR B1 0401, 0404 and 0101, referred to as the shared epitope. Smoking may trigger protein citrullination leading to shared epitope-restricted autoimmunity resulting in the generation of ACPA.3
With reference to these classical biomarkers, the typical RA presentation combines inflammatory arthritis with the presence of rheumatoid factors, ACPA and the HLA shared epitope.4 These markers are clearly associated with a high risk of joint destruction (figure 1). It is likely that some aspects of the pathogenic process of RA are different in the presence versus the absence of these factors, and even though the disease course is generally more severe in the presence of several of the determinants mentioned above, major destruction can still be seen in their absence.

Pathogenesis of rheumatoid arthritis (RA) subsets according to the presence of anticitrullinated protein antibodies (ACPA).
Overall, RA appears now to be a syndrome with different mechanisms underlying different clinical subsets and there is a need for better biomarkers for these new subsets at the three critical time points.5 We will now consider the various characteristics required for a biomarker for diagnostic, prognostic and treatment purposes.
Biomarkers in the preclinical and early phases of RA
This is clearly a situation where any improvement, even a delay in the start of the disease, will have major benefits at the individual and societal levels. Acting early is the best way to prevent destruction. Moreover, it is much easier to achieve remission in symptomatic patients if treatment is started early.
Diagnostic tests to identify preclinical RA when clinical manifestations are not yet present and prognostic tests to predict progression are needed to establish the rules for early intervention. A new set of criteria has been introduced for the diagnosis of RA, taking into account the use of ACPA.6 The next step is the identification of markers to distinguish undifferentiated arthritis from RA. The identification of biomarkers at this stage and the development of tools combining markers and stage-related clinical characteristics will impact on treatment initiation, selection and duration.
Studies on preclinical samples have indicated that ACPA and inflammatory cytokines are present years before the clinical onset of disease.7 However, no therapies have been evaluated for this early indication and as the prediction algorithms are still incompletely evaluated, trials aimed at prevention should be cautiously designed.8 Indeed, in the absence of clinical arthritis in ACPA positive patients, the synovium is still similar to that of controls.9
Genetic markers remain of limited value, and their use at the preclinical stage could raise ethical issues. Used alone they are unlikely to be sufficiently predictive of disease to warrant population screening. However, they may have a role in the future in quantifying risk in groups already at higher risk of disease, such as those with a family history and other predisposing factors.10
Biomarkers of prognosis and stratification according to baseline risk
The chronicity and activity of local disease have a major effect on joint function and daily life. At the population level, there is an overall positive correlation between joint inflammation and the resulting joint destruction. This is most obvious during the first 2 years of disease. At the individual level, however, the correlation between inflammation and destruction is rather poor.
New prognostic tests to predict the progression of early disease need to be developed to establish rules for intervention. The proper timing at this stage creates a so-called ‘window of opportunity’. Many studies have shown that rapid initiation of effective therapy in early RA leads to better long-term disease outcome.11 Control of destruction is itself a major achievement, but even at the early stage it is still unclear how repair activity can be induced. RA is clearly characterised by an imbalanced destruction/repair activity ratio, as reflected by differences in matrix breakdown products between destructive and non-destructive RA.12
In addition to the effect of local disease, we have to take into account the effects of chronic inflammation on life expectancy and quality of life. The systemic nature of RA-related inflammation has been under-recognised, but it is now widely accepted that the systemic effects of chronic inflammation contribute to cardiovascular events.13 Infections are the other leading cause of death in RA. Here again, chronic inflammation induces immune defects with a negative inhibitory effect on cell mediated immune defences.14 Control of inflammation is a way to reduce these related organ defects, increase survival and improve health-related quality of life.
Biomarkers during treatment
In the past few years, results from clinical trials and clinical observations have established new goals for RA treatment. The major key phrases defining the new concepts of RA treatment are: as early as possible, with a combination of drugs, and with the tightest possible control. The current goal is early and aggressive intervention to seek clinical remission in high-risk patients. Our therapeutic goal should be a more robust clinical response than the partial response defined by the American College of Rheumatology 20 criterion. The use and duration of a placebo arm have to be better justified.2 We should focus on long-term remission based on optimal, individually tailored treatment. In addition, patient safety is crucial to ensure that a therapeutic intervention has the best benefit–risk profile.
Targeted treatment
RA is a very good example of a condition where targeted treatments have led to major progress. This was first observed with a tumour necrosis factor (TNF) inhibitor combined with methotrexate. However, we still do not select patients based on specific evidence of activation of TNF pathways. This is largely due to the lack of biomarkers that define patients with more or less dependence on TNF-dependent pathways, and thus the lack of trials to verify the prognostic value of such biomarkers.15 There are arguments to suggest a better response in patients with increased local and systemic levels of TNF or synovial tissue expression.16 17
A simple classification of biomarkers could consider their drug-targeted or non-targeted nature. Drug-targeted biomarkers are specific for a target such as TNF and would be particularly suitable for a disease or a clinical subset where highly targeted therapies are available. In contrast, non-targeted biomarkers are common for disease parameters, for various treatment endpoints, for example, destruction or inflammation.
Safety issues
The new rules of RA treatment could also address safety issues. As example, multiple cases of tuberculosis were observed with the initial use of TNF inhibitors in routine practice. As a rule, these rare events are not detected in clinical trials. Therefore, identification of new safety biomarkers would require collaboration to collect sufficient numbers of sample. As sample collection would probably only be feasible retrospectively, we should look for a stable biomarker, such as genetic markers, to predict these rare adverse events. Such markers would need to have a large effect to be detectable in small sample numbers. There is however precedent for this,18 for example, liver toxicity from flucloxacillin and HLA-DRB*5701 (OR >80),19 or thiopurine S-methyltransferase gene polymorphism and azathioprine-induced bone marrow suppression. In contrast to genetic studies of complex phenotypes like susceptibility to a certain disease where sample sizes have to be in the hundreds to thousands to reach significance, for rare, well-defined adverse events, smaller sample numbers (typically 50–100) are required.
Major safety issues with biologics remain a real concern and include the risk of cancer and severe infections, often in the context of complex immunosuppression (tuberculosis, progressive multifocal leukoencephalopathy, etc).20 21 Improvement of early signal detection of safety issues is needed to allow implementation and ongoing monitoring of risk minimisation measures. Collection of samples from these patients is mandatory to improve understanding and prevention.
Treatment management
Methotrexate and biologics are commonly prescribed at a fixed dose or at best according to body weight. Monitoring the bioavailability of biologics via pharmacodynamic biomarkers is just starting. Although drug trials focus on response rates, the definition of non-response is at least as important. The goal is to avoid patient exposure to a drug without the anticipated benefit. In addition, the cost-effectiveness of a drug depends on results in non-responders. The type of unresponsiveness depends on the duration of drug exposure. Short-term response is achieved within the usual duration of clinical trials (6 months) and refers to primary response. Long-term response includes remission with absence of clinical symptoms and loss of response with time (eg, antidrug antibodies).22
With the use of new biomarkers, evaluation of therapy could be based on a combination of predictive value, efficacy and safety. Today, switching between treatments occurs because of lack of efficacy or for safety issues. In the future, switching could be justified because of the need to achieve different targets, for example, induction of repair instead of control of inflammation. Accordingly, sequential treatment could start with control of inflammation followed by means to reinduce an efficient regulatory pathway, for instance by targeting T cell functions.
More targets and more tools
The future of RA care is in transition with better understanding leading to a large number of new targets. In parallel, progress in technology has now transferred to the clinic resulting in a huge volume of results on the heterogeneity of genes, RNA transcripts and proteins between patients.
More targets
Over the last 20 years, the number of drugs registered for RA has increased to the point that there are now five anti-TNF drugs on the market. Other interleukin (IL) pathways and T and B cells are also being targeted with specific tools. These biological therapies have been a major advance in both achieving clinical responses and preventing/retarding bony erosions and joint space narrowing. These specific, more potent molecules are more commonly used in combination with other agents, such as methotrexate, in standard care. In order to develop new therapies, initiatives are focused on the development of new biological agents and small molecules directed at signal transduction pathways. We will need to evaluate these new molecules either in combination with the available biologics, or in patients for whom the latter are not suitable.
With multiple important molecular targets (eg, TNF, IL-1, IL-6, IL-17, and many others), RA is now seen as a clinical syndrome with subsets linked to different pathogeneses. Specificity of therapy may depend on knowing which target is playing the key role in the clinical manifestations in a particular patient at a particular stage of the disease.
More tools
High throughput genomic, transcriptomic, proteomic and metabolomic technologies are increasingly being applied to clinical samples. These technologies have been a major revolution regarding the quantity and quality of information obtained from clinical samples.23 As an example, genome-wide analysis has confirmed major differences between ACPA-positive and ACPA-negative disease.24 Transcriptomic analysis generates useful information for the identification of new pathways that facilitates the search for biomarkers and new targets for treatment.25 26
Routine access to the synovium
The synovial membrane is the primary site of inflammation in RA and is highly heterogeneous at both the cellular and the molecular level. Thus, as in other conditions such as lupus nephritis and cancer, the role of synovial tissue biopsy as a tool to stratify patients into different prognostic and responsive groups has been explored.
Advances in novel methodologies such as minimally-invasive ultrasound guided synovial biopsy and digital image analysis have made the acquisition and investigation of synovial tissue much easier.27 28 However at the moment, treatment decisions are not yet routinely based on synovium analysis, as would be the case for lupus nephritis. Several prognostic pathological markers have been reported. For example, sublining macrophages have been shown to reliably correlate with response to therapy and, unlike some of the clinical measures (eg, disease activity score in 28 joints), they do not change with ineffective therapy.29 A number of collaborative initiatives such as the European Synovitis Study Group linked to the OMERACT network are currently ongoing to identify novel prognostic biomarkers and investigate the clinical utility of synovial pathobiology in disease evolution, outcome and response to therapy.5 30
Application of the concept of personalised medicine to RA
The public and private sectors are now both looking at the use of personalised medicine to provide better care.31 One of the first applications of personalised medicine was for breast cancer,32 where identification of molecular targets inside the tumour tissue is now mandatory for the use of targeted treatments. This practice has reached the regulatory level and the payers. In this context, drug trials are ongoing based on targeted biomarkers. Some of these aspects can be applied to RA, where access to the affected tissue is also possible.30 Similar concepts apply also to osteoarthritis.33 Ideally a biomarker in a readily available biosample, such as peripheral blood, would be preferable, although this compartment may not have direct implications for disease pathogenesis. This consensus has identified key issues to be clarified for biomarker discovery, registration and routine use (box 1).
Box 1 Key issues to be clarified for biomarker discovery, registration and routine use
▶ Access to sample collections, including those stored by drug companies
▶ Location of collections
▶ Financial support for collections
▶ Intellectual property aspects
▶ Collections of samples during the early phase of marketing of new drugs (safety issues)
▶ Price of companion biomarker included in that of the drug
Which type of assay?
It is doubtful that a single biomarker will be able to clarify the large number of questions related to the different subsets of RA. The future seems to lie in the combination of different markers with classical clinical features. Integration of these clinical and biological data from different sources implies the development of appropriate open-use software and its multi-centre validation. The design of clinical studies should allow regulatory requirements to be met and health technology assessments to be carried out.
Reimbursement of biomarker determination is a health policy issue as regards balancing the high cost of biologics. Four of the 10 drugs with the highest cost worldwide are biodrugs with RA as an indication (http://www.in-pharmatechnologist.com/Industry-Drivers/Bio-drugs-to-dominate-top-ten-list-by-2014). Optimal use of biomarkers may lead to a more rational use of these costly treatments. For the payer, the issue is not much different from the cost of ACPA or shared epitope determination. Integration of a multi-parameter calculation can help define the cost-effectiveness of biomarkers.
Sample collection
Collections of biosamples are key for the identification of biomarkers. Because of the efforts needed to establish and maintain a collection, an important issue is from which patients should samples be collected: all, those receiving biotherapies, those enrolled in clinical trials, etc. Ideally, samples from patients on new treatments should be collected, particularly if rare and unpredictable events are being studied or are expected. The second aspect is the nature of the samples (serum/plasma/urine, DNA, RNA, biopsies) and for how long sample collection is required. In that respect, standardised protocols for sampling, collecting and processing are needed. This is particularly critical for mRNA and tissue biopsies, and not so much for DNA.
Samples for collections are obtained under ethics control. In order to build multicentre interactions, ethical rules should be set at the EU, and not at a national, level. Because of the long time span needed for collecting, collections have not been adequately supported financially. Various options for support are possible: the health system itself, the price of the drugs to include the cost, research funding, drug industry, etc. For current biologics at least, funding by health systems should include support for collections. Finally, these collections are a source of intellectual property and this has to be clarified early.
Infrastructure
The location where collections should be kept (hospitals or central facilities) has not yet been clarified. Drug companies would like to control collections obtained during their own clinical trials but formalised sample access procedures are needed. At this stage, access to these collections for academia has to be facilitated. Ethics committees should agree on the use of samples obtained for trials by third parties.
Molecular diagnostics laboratories have to be certified to carry out diagnostic testing. The validation and qualification phases of diagnostic testing should be carried out in certified laboratories or service centres. The degree and qualifications of the individual in charge of such tests have not yet been clearly defined. Similar to the title of ‘responsible pharmacist’ for the drug industry, that of ‘responsible biologist’ has been proposed for the diagnostics industry.
The huge amount of data that will be generated with the option of pooling data sets from industry and consortia requires centralised data storage facilities where data can be collected and mined.
Validation, qualification and registration of biomarkers
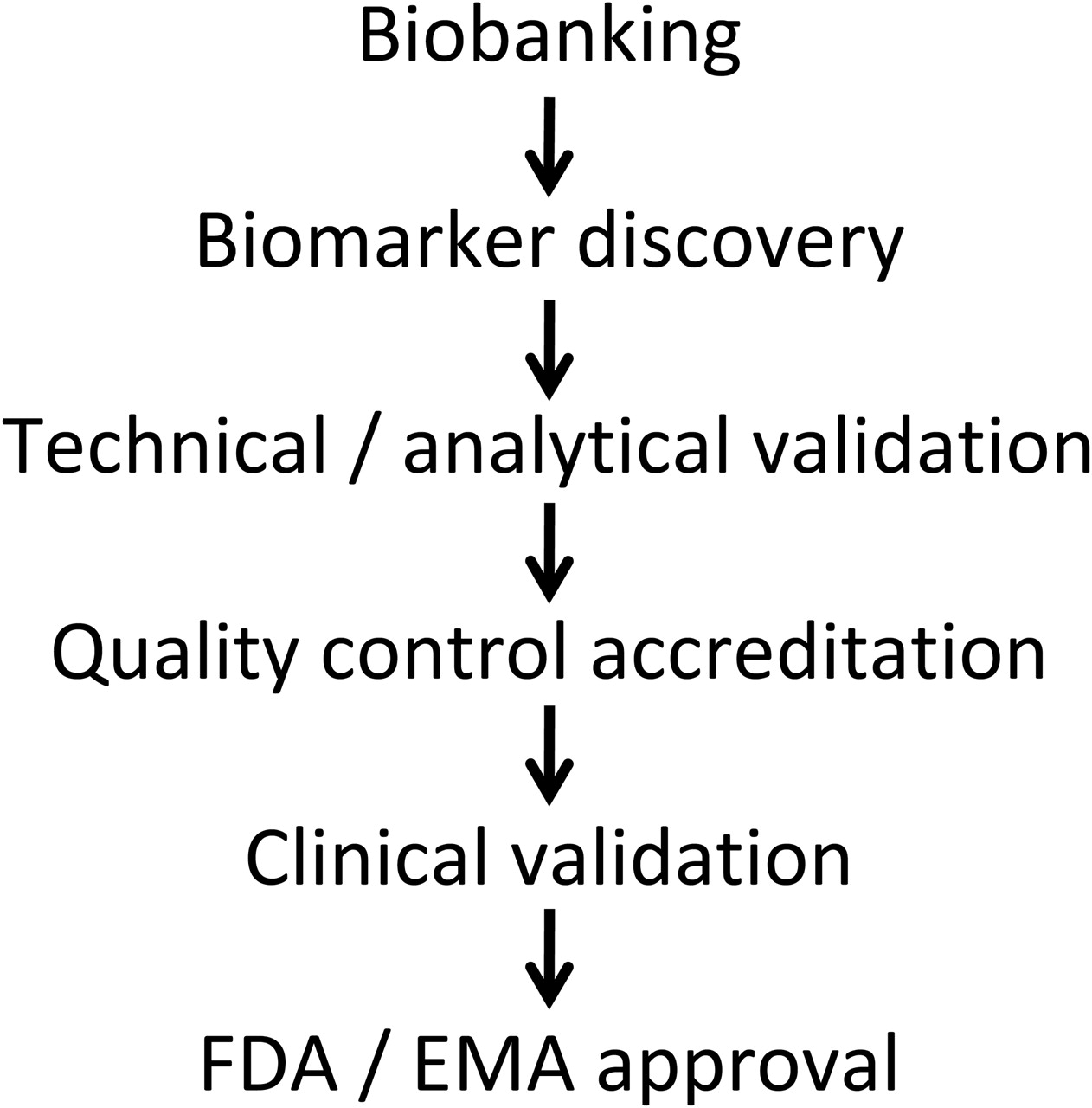
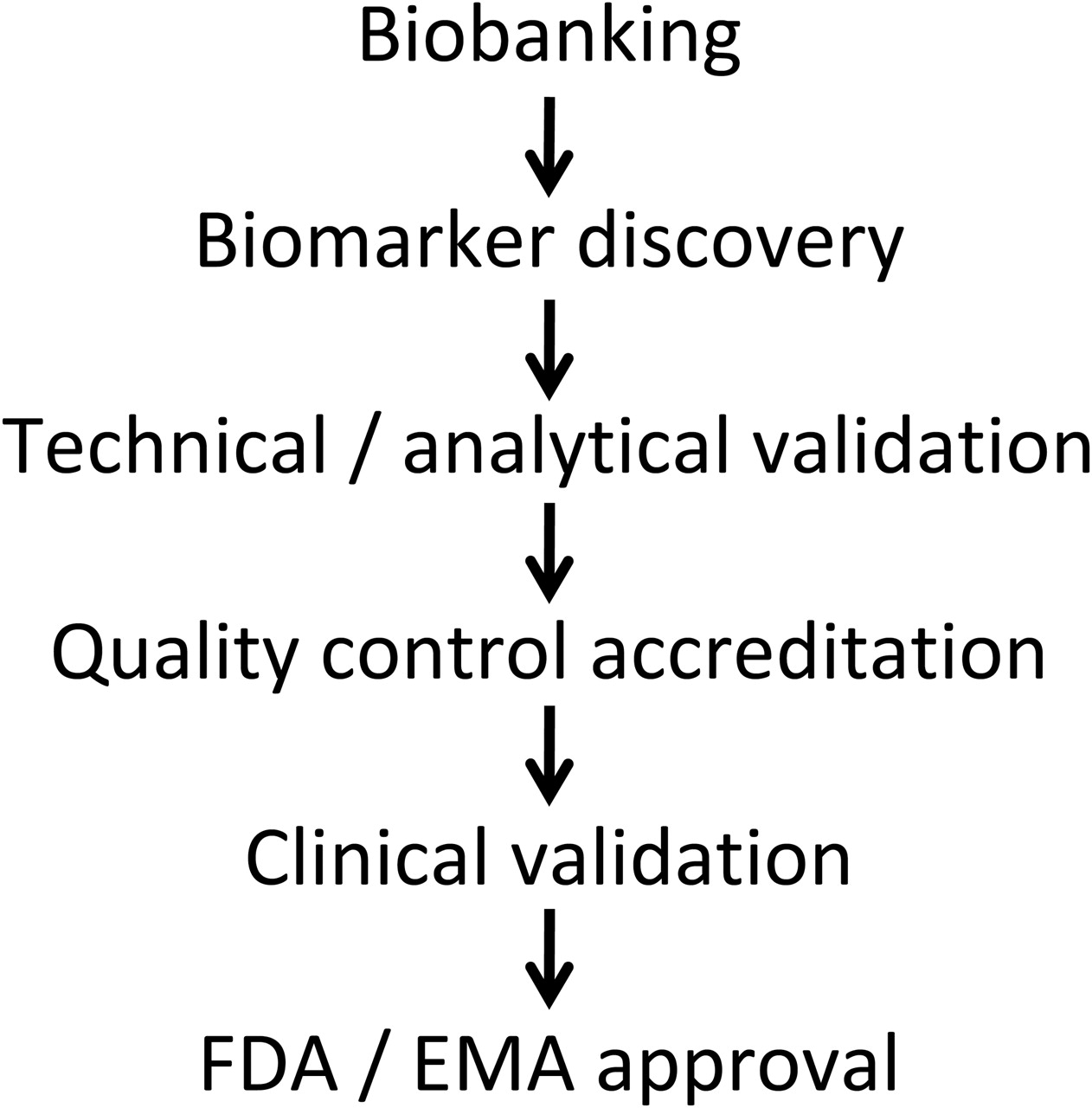
The agreement of EU and US regulatory authorities is required before a biomarker can be effectively used. The first step is to define and establish validation cohorts with biosamples, for example, within existing consortia (figure 2). The quality of the biomarker will reflect the quality of the collection. Accordingly, there is a need for an inventory of possible sources of validation cohorts, samples already available and the ethics around their use.
{kind=link}
{kind=link}
Steps in biomarker development and registration. See text for details and differences between the Food and Drug Administration (FDA) and the European Medicines Agency (EMA).
The next step is technical and analytical validation followed by quality control and accreditation of the assay. The successive processes of biomarker development include two major steps, the validation and qualification phases. The validation phase involves assessment of the assay or measurement of performance characteristics including sensitivity, specificity and reproducibility. The qualification or evaluation phase is the evidentiary process linking a biomarker to a clinical endpoint. According to the Food and Drug Administration (FDA), this phase comprises two levels, the ‘probable’ valid biomarker process and the ‘known’ valid biomarker process. Whereas the ‘probable’ biomarker has not yet received the necessary scientific scrutiny, the performance characteristics of the ‘known’ valid biomarker have reached widespread agreement. As discussed previously, the steps will include the first retrospective validation of collected biosamples followed by a prospective validation. The final step will be acceptance by European and US regulatory authorities.
Diagnostic tests follow different regulatory processes in the EU and US. In the EU, there is no official EU regulatory approval process for in vitro diagnostic (IVD) tests. Formally, they can be described as medical devices, which will be accepted by a notified body if they comply with certain generally accepted ISO standards. In the US, the FDA has also developed a regulatory process for IVD devices and biomarker qualification to accelerate the process by which new biomarkers are integrated into the development of therapies.34 Multiplex IVD devices are referred to as ‘in vitro diagnostic multivariate index assays’. As is true for all medical devices, regulatory classifications are determined by intended use(s) and device risk. The FDA draft guidance also addresses premarket pathways and postmarket requirements. Therefore, the regulatory aspects are best approached through a constant interaction with the competent authorities. The European Medicines Agency (EMA) and the FDA have groups dedicated to the development and use of biomarkers. The FDA Voluntary Genomics Data Submissions and the EMA Briefing Meetings provide non-committing, secure environments to explore the regulatory process for biomarker qualification. Currently health authorities are working on a guidance for an international harmonisation document on the biomarker qualification process. Recently, joint EMA–FDA activities in biomarker qualification became part of the confidentiality arrangements between these two authorities. National regulatory authorities within the EU contribute via their delegates/experts to discussions at the EMA level.
There are various options for the timing of validation. Validation during clinical trials only would allow the identification of a companion biomarker. Validation during postmarketing/routine use is best suited to rare events and safety issues. It also allows studies on long-term response in a real-life situation. Finally, the other option is validation performed during a trial based on biomarkers. Possible differences in regional and ethnic populations should be carefully considered.
Registration of a drug with a companion biomarker
Although this option has been highly advertised, there are only a few successful examples. The FDA has a special office for companion diagnostics and has released a concept paper on ‘Early Development Considerations for Innovative Combination Products’ (http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126054.pdf). A fear of the industry is the possible limitation of market size with a marker to select a subset of patients. The key advantages, however, are clear, providing an optimal balance between the number of treated patients (responders and/or improved safety) and duration of treatment (better compliance).
Trials based on biomarkers
These biomarkers will also be used to select patients for inclusion in trials. There are already examples of the use of biomarkers in the official indication of drugs for certain cancers.35 Although the acceptance of biomarkers as clinical efficacy or safety endpoints can be seen as the ultimate goal for RA, we are not yet there. As a first step, we could still use biosamples collected during trials and study whether the conclusion would have been different if the trial had been based on a particular marker. Another aspect is the validation of biomarkers as surrogate endpoints. Even if a biomarker is not completely validated as an accepted surrogate endpoint, these markers could be used in proof of concept studies or dose selection.
Here again, the key interactions in a connected partnership are between academia and the drug and biomarker industries. Commonly there is clear separation between the industry developing drugs and the industry developing biomarkers, which implies separate discussions with the authorities. The key issue will be the price of and reimbursement for such companion biomarkers.
Conclusion
RA is one of the most appropriate conditions for the application of personalised medicine as a high degree of heterogeneity has been recognised, which remains to be explained. Such heterogeneity is also reflected in the large number of treatment targets and options.
In order to make the best use of these treatment options, targeted and non-targeted biomarkers have to be identified and validated. To this aim, new rules are needed for the interaction between academia and industry under regulatory control. Setting up multi-centre biosample collections with clear definition of access, organising early, possibly non-committing discussions with regulatory authorities, and defining a clear route of validation, qualification and registration of the biomarker–drug combination are some of the critical steps where effective collaboration between the drug industry, academia and regulators is needed.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.