Article Text

Abstract
Objective: To assess the efficacy and safety of leflunomide in active ankylosing spondylitis (AS) compared with placebo in a 24 week pilot study.
Methods: In a single centre randomised, double blind, placebo controlled study, 45 patients with active AS were randomised to either leflunomide 20 mg daily (n = 30) or placebo (n = 15). Active disease was defined as a score of ⩾4 on the Bath ankylosing spondylitis disease activity index (0–10), and pain of ⩾4 on a visual analogue scale (0–10). The primary efficacy variable at week 24 was the 20% response rate, as recommended by the Assessments in Ankylosing Spondylitis (ASAS) working group. Secondary outcome variables included general wellbeing, metrology index, swollen joint count, erythrocyte sedimentation rate, and C reactive protein.
Results: In all, 13 women and 32 men were studied. Demographic and disease indices were comparable between the two treatment groups at baseline. The rate of ASAS 20% responders was not significantly different: 27% in the leflunomide treated patients and 20% in the placebo group (95% confidence interval, −32% to 19%). No significant differences were found between the treatment groups in mean changes of the secondary outcome variables. Eleven patients were withdrawn prematurely from the study because of adverse events (7), lack of efficacy (3), and non-compliance (1). Most frequently adverse events were gastrointestinal side effects and skin disorders.
Conclusions: In this placebo controlled study, leflunomide treatment did not result in a significant improvement of the ASAS 20% response in active ankylosing spondylitis. No unexpected or severe adverse events occurred.
- AS, ankylosing spondylitis
- ASAS, Assessments in Ankylosing Spondylitis
- BASDAI, Bath ankylosing spondylitis disease activity index
- BASG, Bath ankylosing spondylitis global score
- BASMI, Bath ankylosing spondylitis metrology index
- DMARD, disease modifying antirheumatic drug
- MMP, matrix metalloproteinase
- NSAID, non-steroidal anti-inflammatory drug
- SJC, swollen joint count
- VAS, visual analogue scale
- disease modifying antirheumatic drugs
- ankylosing spondylitis
- leflunomide
Statistics from Altmetric.com
- AS, ankylosing spondylitis
- ASAS, Assessments in Ankylosing Spondylitis
- BASDAI, Bath ankylosing spondylitis disease activity index
- BASG, Bath ankylosing spondylitis global score
- BASMI, Bath ankylosing spondylitis metrology index
- DMARD, disease modifying antirheumatic drug
- MMP, matrix metalloproteinase
- NSAID, non-steroidal anti-inflammatory drug
- SJC, swollen joint count
- VAS, visual analogue scale
Ankylosing spondylitis (AS) is an autoimmune disease characterised by chronic inflammation of the sacroiliac and spinal joints and entheses. The disease occurs mainly in young adults and can lead to stiffness and deformity of the vertebral column, with invalidating deformities. AS is often accompanied by extraspinal manifestations as arthritis of the peripheral joints, and involvement of the eye (acute anterior uveitis), heart, and lungs.
Treatment was, until recently, mainly based on non steroidal anti-inflammatory drugs (NSAIDs) and physical therapy.1 The disease modifying antirheumatic drugs (DMARDs), for example sulfasalazine and methotrexate, seem to be less beneficial in AS than in other rheumatic diseases such as rheumatoid arthritis.2 Recent studies have shown that the tumour necrosis factor α (TNFα) blocking agents infliximab and etanercept are very effective in a large proportion of patients with AS.3,4 These powerful drugs, however, are costly and are sometimes accompanied by severe adverse effects such as opportunistic infections. Moreover, these agents fail to reach efficacy in approximately 30% of the patients.5 For these reasons we investigated the efficacy and safety of another DMARD, leflunomide—a drug proven to be effective in rheumatoid arthritis.6–,9 In addition, leflunomide shows beneficial effects in patients with psoriatic arthritis which, like AS, belongs to the group of spondyloarthritides.10–,13 In the present study the efficacy and safety of leflunomide was investigated in patients with active AS in a randomised, double blind, placebo controlled trial.
METHODS
Consecutive patients with AS were recruited from the outpatient rheumatology clinic of the Jan van Breemen Institute (referral centre) and the surrounding hospitals in Amsterdam. The study was carried out in the period from March 2002 to September 2003. The study group comprised 45 patients aged 18 to 70 years with active definite AS, diagnosed according to the modified New York criteria.14 Active disease was defined as: at least a 4 point score on the Bath ankylosing spondylitis disease activity index (BASDAI; scale 0–10) and a pain score of at least 4 on a visual analogue scale (VAS; scale 0–10) at screening.
NSAIDs and corticosteroids up to a maximum daily dose of 15 mg prednisone were allowed on an unchanged regimen for at least 30 days before the study drug administration and throughout the study. The use of DMARDs (particularly sulfasalazine, methotrexate, and TNF blocking agents) during the study and within the 30 days before the study was not permitted.
Both men and women were required to practice contraception. In female patients of childbearing potential a urine pregnancy test was done at baseline and any pregnant women were excluded.
Other exclusion criteria were: impaired hepatic enzyme tests (alanine and aspartate transaminases more than twice the upper laboratory limit of normal), impaired renal function (serum creatinine >110 μmol/l), untreated hypertension, malignancy, diagnosis of other inflammatory joint diseases, impaired bone marrow function, recent serious infections, and any clinically relevant cardiovascular, hepatic, neurological, endocrine. or other major systemic diseases.
Design
Forty five eligible patients were randomised at baseline (within four weeks after screening) to receive either leflunomide (n = 30) or matching placebo (n = 15) for 24 weeks. The dosage schedule included a loading dose of 100 mg (given on day 1, 8, and 15) and leflunomide 20 mg on the other days during the first three weeks, followed by a maintenance dose of 20 mg daily. We often apply this slightly modified dosing schedule, as we have the impression that the rate of gastrointestinal complaints is less than with the standard schedule. Compliance was assessed by tablet counts of returned study preparations and was calculated on the basis of the number of days in the study.
Efficacy and safety indices (vital signs and adverse events) were assessed at baseline, week 4, week 12, and week 24. The following efficacy variables were used: BASDAI,15 pain assessed on a VAS (0–10), BASFI,16 Bath ankylosing spondylitis global score (BASG),17 Bath ankylosing spondylitis metrology index (BASMI),18 44 swollen joint count (SJC), general wellbeing on a VAS (0–10) according to the physician, erythrocyte sedimentation rate (ESR), and C reactive protein.
Blood tests (haematology and blood chemistry) were monitored every two weeks during the first six weeks and at regular intervals later during the study period.
Statistics
Based on an assumed 50% response rate for leflunomide and 20% for placebo, adjusting for a non-evaluable rate of 25%, 61 patients per treatment would have been needed to reach a level of significance of 0.05 with a power of 80%. Underlining the explorative character of this trial, and because of obvious feasibility issues in this single centre study, it was decided to randomise 45 patients. Expecting a higher dropout rate in the leflunomide group, a randomisation ratio was defined with a 2:1 balance (30 leflunomide patients and 15 placebo patients) so as to ensure a minimum collection of efficacy and safety data in a new indication for leflunomide. Altogether, the power of the study was reduced to 65%, which reasonably can be said to show at most an effect trend.
The primary efficacy variable was the 20% response rate, as recommended by the Assessments in Ankylosing Spondylitis (ASAS) working group.19 Each patient was classified as a treatment responder or non-responder at the end point (week 24 or last available observation under treatment). The ASAS 20% response was defined as improvement of ⩾20% and absolute improvement of ⩾1 unit (on a scale of 0 to 10) in at least three of the following four domains and absence of deterioration (change for the worse of ⩾20% and net worsening of ⩾1 unit) in the remaining domain: patient global assessment, pain (VAS), BASFI, and inflammation (the mean of the two morning stiffness related BASDAI VAS scores).
The responder rates in the two treatment groups were compared using the binomial test for two samples, and the 95% confidence intervals were calculated.
Subgroup analyses were done in patients with an ESR of >30 mm/h or a C reactive protein of >10 mg/l, or both, at baseline, and in patients with peripheral arthritis (SJC >1 at baseline).
The secondary efficacy variables at end point were compared with the assessments at baseline and 95% confidence intervals for the differences between leflunomide and placebo for the mean changes from baseline were calculated. Student’s t test was used for variables with a normal distribution and the Mann–Whitney U test for variables with a non-normal distribution. A two sided p value of less than 0.05 was considered significant.
The primary and secondary efficacy analyses and safety analysis were carried out on an intention to treat basis.
The efficacy analysis was also done for the per-protocol (PP) population. We defined the PP population as all patients treated with a minimum drug exposure of eight weeks, and excluding one patient in whom the prednisone dose was increased. The PP population (36 patients) was identified before unblinding the treatment allocation.
The study was approved by the local ethics review board, and written informed consent was obtained from all patients.
RESULTS
Baseline characteristics
The demographic data of the patients and disease characteristics at baseline are summarised in table 1⇓. The variables were comparable and not statistically different between the two treatment groups. Fourteen patients had a history of uveitis (eight leflunomide and six placebo). Two patients in the leflunomide group and one in the placebo group had inactive inflammatory bowel disease and one patient in both groups had psoriasis. NSAIDs were used by 93% of the leflunomide and 87% of the placebo treated patients. Corticosteroids were used by one patient. None of the patients had ever used a TNF blocking agent.
Demographic and mean (or median) baseline characteristics (and ranges) of 45 patients with active ankylosing spondylitis
Primary efficacy variable
The number of responders according to the ASAS 20% definition was 11: 8 (27%) in the leflunomide group and 3 (20%) in the placebo group (95% confidence interval (CI) for difference, −32% to 19%).
In the subgroup with an ESR of ⩾30 mm/h, or a C reactive protein of ⩾10 mg/l, or both, at baseline (n = 26), the response rate was 32% and 29% for the leflunomide and the placebo group, respectively (−42% to 36%).
In the subgroup of patients with peripheral arthritis (SJS ⩾1 at baseline), which included only four patients, the response rate was 50% in the leflunomide and 100% in the placebo group (−19% to 100%).
In the per-protocol population the response rate was 6/24 (25%) in the patients treated with leflunomide and 2/12 (17%) in the placebo treated patients (−36% to 19%).
Secondary efficacy variables
The mean secondary efficacy variables at baseline and the mean changes after 24 weeks did not show any significant differences of global disease activity (BASG), disease activity index (BASDAI), functional index (BASFI), pain, metrology index (BASMI), swollen joint count or C reactive protein between the treatment groups (table 2⇓). The mean change in ESR was the only variable that differed significantly between the treatment groups in favour of the placebo group. This observation was the result of a skewed distribution of the ESR, because the differences between the median changes were much smaller.
Mean efficacy variables at baseline and the mean change after 24 weeks with the 95% confidence interval for the difference in change between the treatment groups
Adverse events and dropouts
Adverse events were seen in 24 leflunomide cases (80%) and 10 placebo cases (67%). The adverse events were classified as possibly drug related in 18 (60%) and six (40%) of the leflunomide and placebo treated patients, respectively. No serious adverse events were seen. Most frequent or clinically relevant adverse events were: gastrointestinal disorders (57% in the leflunomide and 33% in the placebo treated patients, respectively); upper respiratory tract infections (17% in the leflunomide and 27% in the placebo treated patients); dermatitis and prurigo (13% in both groups); fatigue (in 13% of the placebo treated patients); deep venous thrombosis (in one placebo patient). In the leflunomide group, a rise in liver enzymes to three times the upper normal limit was observed in one patient with borderline liver enzyme tests at baseline. No clinically relevant changes were found in the other laboratory variables. No changes were seen in the mean blood pressures. Antihypertensive treatment had to be increased in one patient treated with leflunomide.
Eleven patients were withdrawn prematurely from the study (fig 1⇓), for the following reasons: adverse events (five leflunomide patients, two placebo patients); lack of efficacy (three leflunomide patients); non-compliance (one placebo patient). Four of these dropouts were responders (three leflunomide patients and one placebo patient). Adverse events leading to withdrawal were gastrointestinal disorders (4), malaise (1), exacerbation of pain (1), headache (1), and erectile dysfunction (1).

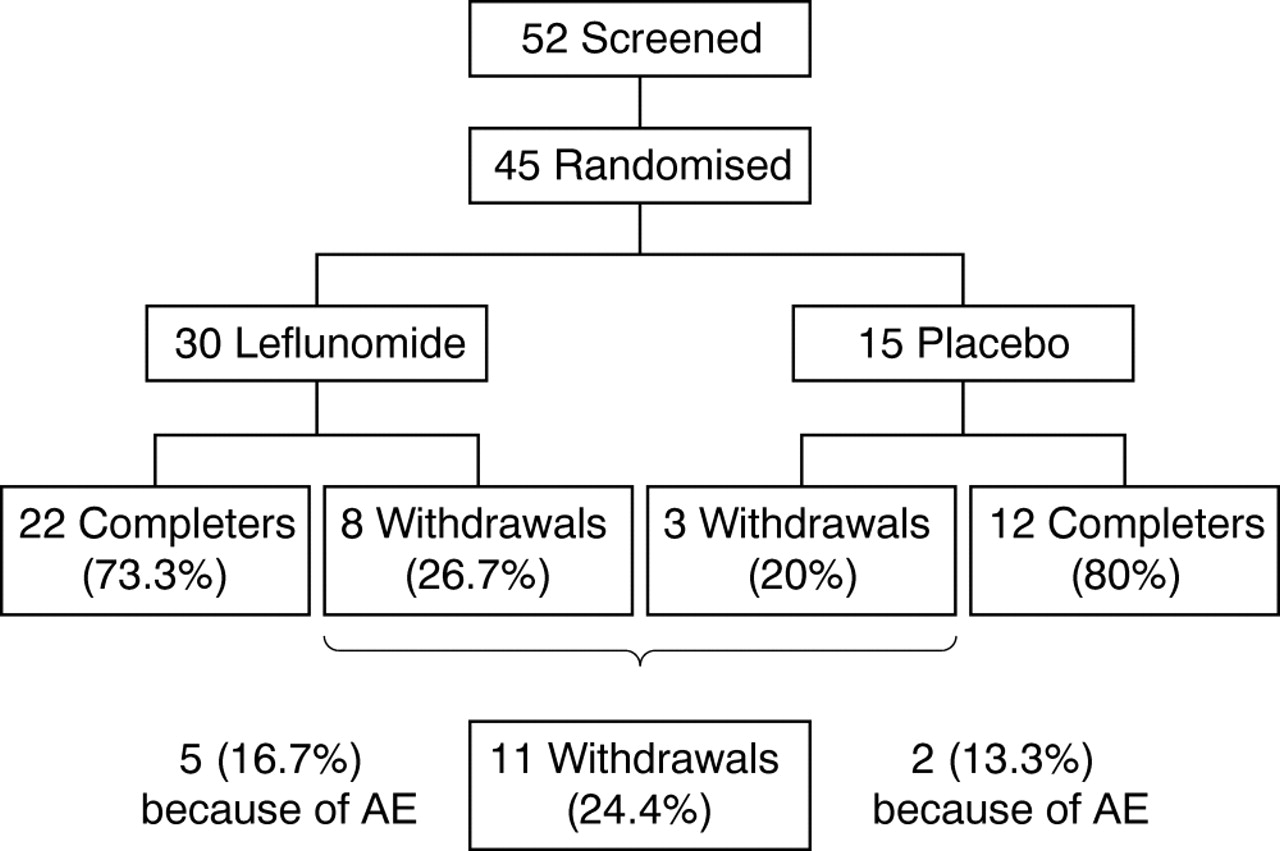{kind=link}
Trial profile. Five screened patients are not included because they did not meet the criteria for disease activity, and two patients because they wanted to have a child. AE, adverse events.
Compliance
On the basis of the number of days in the study (leflunomide, mean 134 days; placebo, mean 142 days) and the number of returned tablets, the calculated compliance was 96% in the leflunomide group and 93% in the placebo group.
DISCUSSION
Leflunomide did not result in significant clinical improvement in active ankylosing spondylitis in this placebo controlled study. The number of AS patients who fulfilled the ASAS 20% response rate was higher in the leflunomide group than in the placebo group, but the difference did not reach statistical significance. The changes in the individual secondary disease outcome indices were comparable. Importantly, no serious or unexpected adverse events were encountered.
Only one other study has investigated the efficacy of leflunomide in AS; this was an open label non-comparative trial of 20 AS patients who were treated for 24 weeks.20 Ten patients were prematurely withdrawn because of lack of efficacy, side effects, or non-compliance. In line with our results, leflunomide seemed to be ineffective. The only significant improvement observed was in a subgroup of 10 patients suffering from peripheral arthritis. In our study, only four patients had peripheral arthritis in at least one joint and therefore we could not draw any conclusions about the efficacy of leflunomide in this subgroup.
The efficacy of leflunomide in AS in our study appears disappointing but it is consistent with the experience with other DMARDs in AS. Most studies with DMARDs in AS, particularly sulfasalazine, show improvement of the peripheral arthritis and ESR, but no significant improvement in the axial complaints.2,21 Mesalazine, which is very effective in inflammatory bowel disease, was not shown to be effective in AS.22 Limited data are available of methotrexate in AS, but a recent small placebo controlled trial (in which a lower dose was used than in most studies in rheumatoid arthritis) showed favourable results.23 The large number of patients with peripheral arthritis in that study (65% of the 17 recruited patients) was striking.
In rheumatoid arthritis leflunomide reduces the numbers of macrophages and T cells infiltrating the synovium, and the expression of adhesion molecules, proinflammatory cytokines, and mediators of joint destruction such as matrix metalloproteinases (MMPs).24 Whereas the molecular pathways have been thoroughly analysed in rheumatoid arthritis, the mechanisms of joint inflammation and destruction are less well understood in AS. In sacroiliac biopsies from 32 patients with spondyloarthritis, T cells and macrophages were the predominant cells, suggesting an important role for these cell types in the pathogenesis.25 Moreover, TNFα blocking treatment in AS downregulates both cytokines secreted by T cells and MMPs in the synovium in spondyloarthritis.26,27 Therefore an effect of leflunomide on AS could be expected. It is interesting that significantly higher serum MMP-3 concentrations are encountered in AS patients with peripheral joint disease than in those with only axial symptoms.27 This latter finding could be the pathophysiological explanation for a possible effect of leflunomide in cases of AS with peripheral joint disease.
It is clear that leflunomide is not effective when compared with the very efficacious TNFα blocking agents, although direct comparative trials have not been conducted. An interesting observation from a trial of anti-TNFα treatment in AS was that patients with a raised C reactive protein had greater benefit from the treatment than those with lower levels.3 However, in our study no difference in responder rate between the treatment groups was seen in the subgroup with an ESR of ⩾30 mm/hour or a C reactive protein of ⩾10 mg/l, or both, at baseline.
Conclusion
This is the first double blind, placebo controlled study of leflunomide in active AS. Unfortunately, it was not possible to show that it had significantly better efficacy than placebo. Whether or not this drug is beneficial for AS patients with peripheral arthritis remains to be established, as in our study the number of such patients was too small to allow us to draw meaningful conclusions. This might be an interesting subject for future research.
Acknowledgments
The study was supported financially by Aventis Pharma, Hoevelaken, Netherlands
REFERENCES
Footnotes
Published Online First 18 May 2005