Article Text

Abstract
Signal transduction pathways regulate cellular responses to stress and play a critical role in inflammation. The complexity and specificity of signalling mechanisms represent major hurdles for developing effective, safe therapeutic interventions that target specific molecules. One approach is to dissect the pathways methodically to determine their hierarchy in various cell types and diseases. This approach contributed to the identification and prioritisation of specific kinases that regulate NF-κB and the mitogen activated protein (MAP) kinase cascade as especially attractive targets. Although significant issues remain with regard to the discovery of truly selective kinase inhibitors, the risks that accompany inhibition of fundamental signal transduction mechanisms can potentially be decreased by careful dissection of the pathways and rational target selection.
- signal transduction networks
- rheumatoid arthritis
- AP1, activator protein 1
- ERK, extracellular regulating kinase
- FLS, fibroblast-like synoviocytes
- IKK, I kappa B kinase
- IL, interleukin
- JNK, c-Jun-N-terminal kinase
- MAP, mitogen activated protein
- MAPKK, MAP kinase kinase
- MAP3K, MAP kinase kinase kinase
- OA, osteoarthritis
- RA, rheumatoid arthritis
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- AP1, activator protein 1
- ERK, extracellular regulating kinase
- FLS, fibroblast-like synoviocytes
- IKK, I kappa B kinase
- IL, interleukin
- JNK, c-Jun-N-terminal kinase
- MAP, mitogen activated protein
- MAPKK, MAP kinase kinase
- MAP3K, MAP kinase kinase kinase
- OA, osteoarthritis
- RA, rheumatoid arthritis
- TNFα, tumour necrosis factor α
In the late 1980s, the cytokine profile of rheumatoid arthritis (RA) was considered so complex and redundant that the notion of anticytokine therapy appeared remote. Through the considerable effort of many investigators, the cytokine network of RA was gradually unravelled. A degree of order was introduced with the recognition that certain cytokines were potential targets for therapeutic intervention. Great consternation ensued when contemplating the possibility of blocking key cytokines because of the potential for untoward or even catastrophic sequelae. Perhaps the price of success in RA would be too high if critical cytokines were inhibited? Perhaps the cytokine network would be so redundant that inhibition of single factors would be doomed to failure?
Of course, we now know the answers to some of these questions. Selective inhibitors of specific cytokines, most notably tumour necrosis factor α (TNFα), were introduced with considerable success. Attention has now turned to other targets in an effort to build on the success of anticytokine therapy. However, these approaches have generally focused on biological agents that are selective for a single factor and must be parenterally administered. More recently, we have directed efforts towards signal transduction pathways because of the possibility of developing oral agents that can regulate the production of an array of cytokine targets. Like cytokine binding proteins introduced several years ago, significant concerns about safety must be considered. The hope is that careful analysis of the signal transduction networks of RA, like cytokine networks, will disclose an organisational hierarchy that can be addressed by highly selective inhibitors.
Multiple signal transduction pathways have been carefully investigated in RA. For instance, elegant studies in rodent models of arthritis suggest that SOCS or JAK-STAT systems can be targeted with gene constructs that express either wild-type or dominant negative proteins.1 NF-κB and mitogen activated protein (MAP) kinases are also attractive pathways for intervention in light of their ability to regulate many genes involved in immune responses.2,3 The enormous diversity of kinases that modulate these and other transduction mechanisms suggests that complex and interrelated events are involved in inflammatory disease.
NF-κB AS A THERAPEUTIC TARGET
Proof of concept studies demonstrating the use of signal transduction blockade in RA are still in their formative stage. However, understanding the regulation of NF-κB is an example of how dissecting the pathway can identify the key proteins that can subsequently be targeted. NF-κB is abundant in rheumatoid synovium, and immunohistochemical analysis demonstrates p50 and p65 NF-κB proteins in the nuclei of cells in the synovial intimal lining.4,5 Although the proteins can also be detected in osteoarthritis (OA) synovium, NF-κB activation is much greater in RA because of phosphorylation and degradation of its natural inhibitor, IkB, in RA intimal lining cells. Nuclear translocation of NF-κB in cultured fibroblast-like synoviocytes occurs rapidly after stimulation by interleukin (IL)1 or TNFα through the activation of I kappa B kinase (IKK) signalling complexes and induces cytokine production. Furthermore, several animal models of arthritis suggest that NF-κB is an attractive target.6
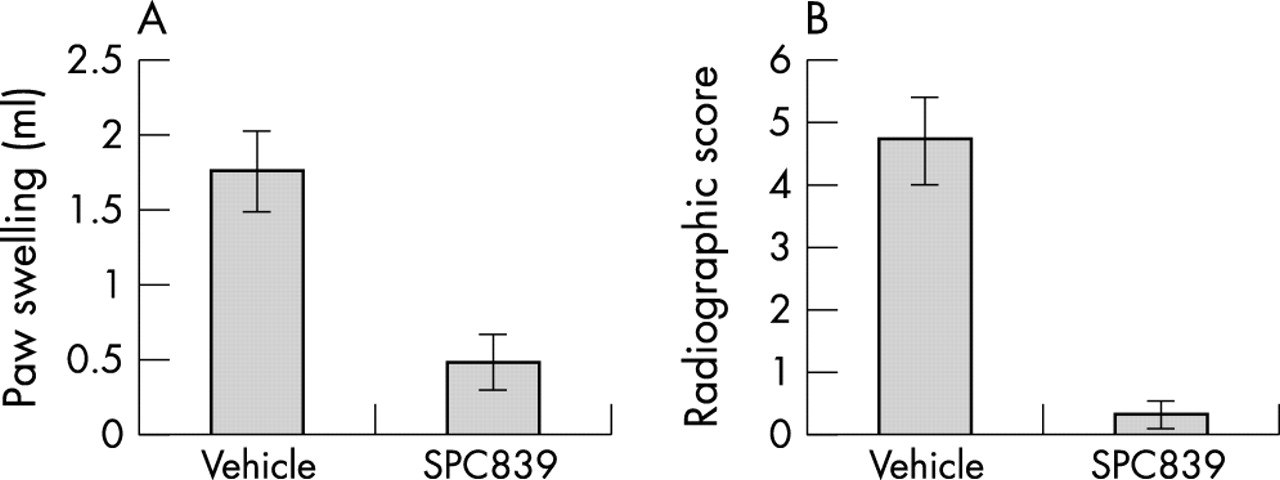
Careful dissection of the NF-κB pathway in RA has focused on a signal complex that includes two IkB kinases, IKK1 and IKK2. Both are constitutively expressed by synoviocytes, and IKK functional activity increases 10- to 20-fold within minutes of cytokine exposure. When dominant negative and constitutively active adenoviral constructs were used, IKK2 was identified as the primary kinase for NF-κB activation in cytokine stimulated synoviocytes. IKK2 blockade in vitro with a dominant negative adenoviral construct inhibits induction of IL6, IL8, and intercellular adhesion molecule-1 (ICAM-1) after stimulation with IL1 or TNFα.7 Intra-articular decoy oligonucleotides or gene therapy using the dominant negative IKK2 adenovirus suppresses joint inflammation in rat models of inflammatory arthritis.8,9 More recently, selective IKK2 inhibitors have demonstrated efficacy in arthritis models (fig 1).10 Hence, careful dissection of the NF-κB pathway in synovial inflammation by several groups identified a specific kinase as a convergence site for cytokine regulation.

Effect of the IKK2 inhibitor, SPC839, in rat adjuvant arthritis. Rats were immunised with complete Freund’s adjuvant and treated with either vehicle or the IKK2 inhibitor SPC839 (100 mg/kg/day beginning on day 15). The experiment was ended on day 28. Paw swelling (A) was determined by water displacement plethysmometry and radiographic damage (B) was determined using a semiquantitative scoring system (maximum score=6). Clinical arthritis and radiographic damage were significantly decreased by the IKK2 inhibitor. Data adapted from reference 10.
A major concern with NF-κB directed therapy is that this particular signalling mechanism participates in many cellular functions, especially those involved with host defence. Blockade could potentially impair both innate and adaptive immune responses.11 NF-κB activation also prevents apoptosis in many cell lineages and raises the issue of enhanced (and unwanted) cell death.12 Although NF-κB inhibition has great potential, balancing the risks and benefits will be crucial. One can take some solace in the fact that several commonly used therapeutic agents might partially interfere with NF-κB, including corticosteroids, leflunomide, sulfasalazine, and even salicylates.13,14
As an alternative approach, new pathways might be identified that regulate only pathological NF-κB activation. For instance, a new inducible IKK known as IKKi was identified in macrophages and can phosphorylate either IkB or IKK2.15 It is constitutively expressed in fibroblast-like synoviocytes (FLS) and can be further induced and activated by proinflammatory cytokines.16 IKKi is also expressed by RA synovial tissue and can be localised to the synovial intimal lining by immunohistochemistry (unpublished data). Although the physiological function of IKKi remains to be defined, it is an example of a signal transduction mechanism that might operate primarily in chronic inflammation rather than homoeostasis.
MAPPING OUT THE MAP KINASE PATHWAY
The MAP kinases represent another attractive target for RA because they can regulate cell proliferation, apoptosis, cytokine expression, and metalloproteinase production. They represent a complex, interrelated signal transduction mechanism that integrates extracellular stresses and induces an appropriate cellular response (fig 2). The three major MAP kinase families, c-Jun-N-terminal kinase (JNK), extracellular regulating kinase (ERK), and p38 kinase, differ in their substrate specificity and subsequent responses to stress depending on the cell type and the environmental influences.17 The MAP kinases regulate various genes through both transcriptional and post-transcriptional mechanisms.18,19 The upstream MAP kinase kinases (MAPKK) serve as regulators of MAP kinase activity by phosphorylating specific threonine and tyrosine residues. MAPKKs are, in turn, regulated, by MAPKK kinases (MAPKKK or MAP3K).20
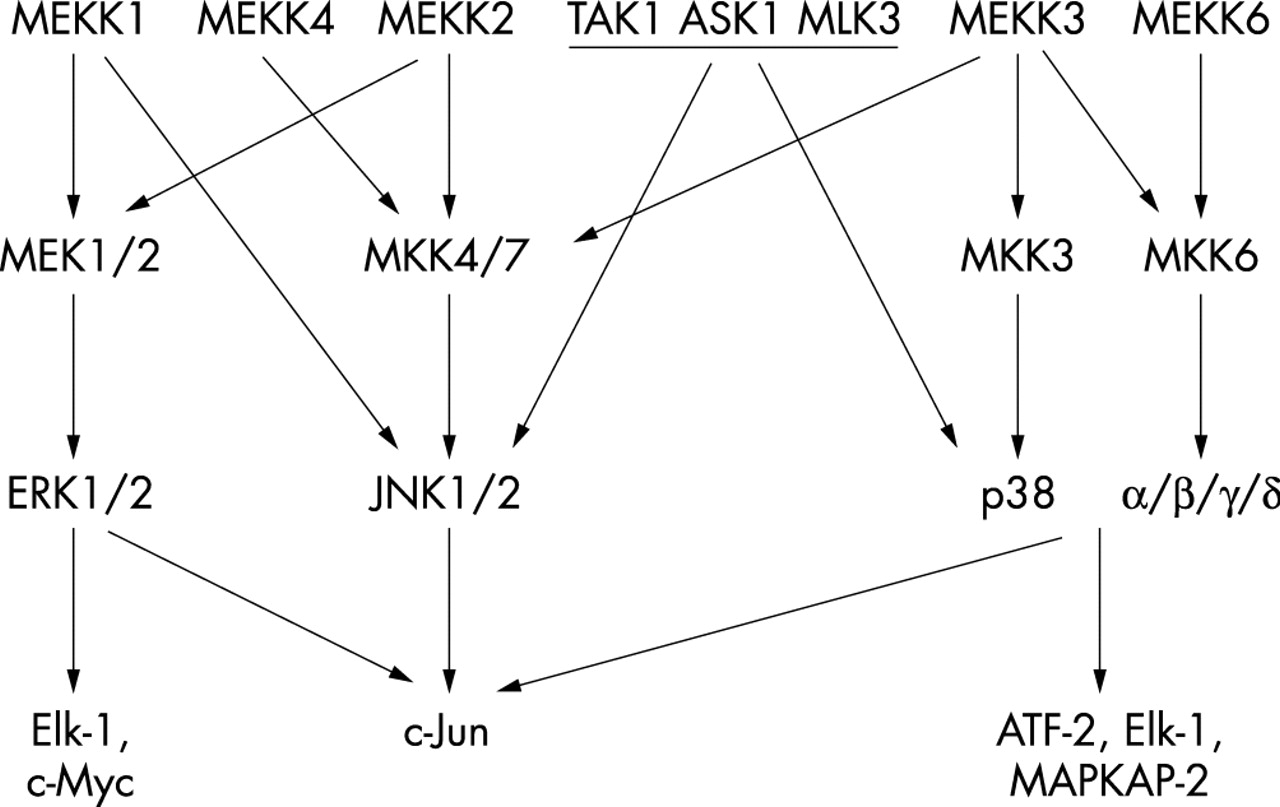
MAP kinase pathways. Complex parallel and interlocking signalling cascades link the three main MAP kinase families, ERK, JNK, and p38. The top level shows the MAP kinase kinase kinases (MAP3Ks), and the second tier shows the MAP kinase kinases (MAPKKs). MKK4 can also activate p38 under some circumstances.
ERK, JNK, and p38 are expressed in synovial tissue and the active phosphorylated forms can be detected by immmunohistochemistry and western blot analysis in RA.21,22 They are also expressed by cultured fibroblast-like synoviocytes, and treatment of cells with proinflammatory cytokines leads to rapid, albeit transient, activation. After stimulation, the cascades continue to expand either by phosphorylation of additional kinases (like MAPKAP-2) or other transcription factors. Cytokine gene expression, especially IL1 and TNFα, are induced by p38 through both pre- and post-transcriptional mechanisms. One of the primary substrates for JNK is c-Jun, which can induce metalloproteinase gene expression by forming homo- and heterodimers and binding to the activator protein 1 (AP1) binding site in the protease promoters.23 The other MAP kinases can also regulate metalloproteinase production, although the relative contributions depend on the specific cell and type of stimulus.
The p38 MAP kinase is of particular interest and several inhibitors have progressed into clinical trials. There are at least four isoforms of p38 (α, β, γ, δ), although the α form is probably the most important in macrophages for cytokine regulation.24 Many of the cytokine regulatory effects appear to be mediated through the downstream p38 substrate MAPKAP-2. Some evidence suggests that TNF production in particular is regulated by this kinase.25 Preclinical models demonstrate that p38 inhibitors are effective in a number of animal models of arthritis, including murine collagen induced arthritis.26 Of note, p38 inhibitors also suppress joint destruction in these models, perhaps by a combination of indirect effects on cytokine expression and a direct effect on metalloproteinase production.
Proof of concept studies demonstrating a biological effect in humans are currently limited to acute administration of p38 inhibitors when endotoxin is given to normal subjects. Pretreatment with a p38 inhibitor suppresses fever and cytokine production (for example, IL6, TNFα, and IL8) up to 90% after oral administration of a single dose.27 Of course, the relevance of endotoxin induced cytokine expression to RA is uncertain. Thus, the role of p38 as a potential therapeutic target in RA can be summarised as follows: (a) p38 is expressed and activated in RA synovium; (b) p38 inhibitors are effective in rodent arthritis models; and (c) p38 inhibitors block cytokine production by macrophages in vitro. However, the primacy of p38 as a regulator of cytokine production in RA remains theoretical. Concerns about toxicity suggest that a selective agent for p38α or perhaps a downstream target might also be desirable.
Although p38 is thought to have an especially important role in cytokine expression, JNK is a key MAP kinase involved in the induction of metalloproteinase genes in RA synoviocytes. Low concentrations of the selective p38 inhibitor SB203580 that completely inhibit p38 function have little or no effect on IL1 induced collagenase expression, AP1 activation, or c-Jun expression.28,29 PD98059, which inhibits MEK1/2 and blocks ERK activation, also modestly decreases collagenase mRNA accumulation. Three isoforms of JNK exist (JNK1, JNK2, and JNK3), and each has multiple alternatively spliced subtypes. Of the myriad of JNK proteins, JNK2 is constitutively expressed by RA synoviocytes and is the primary isoform in this cell type. JNK2 accounts for perhaps 90% of JNK protein in cultured FLS, with the balance being JNK1.
When a JNK inhibitor that blocks all three JNKs was used, marked protection of bone destruction was seen in the rat adjuvant arthritis model together with decreased synovial AP1 activation and collagenase-3 gene expression.29 The question whether a JNK2-specific inhibitor might be useful was examined in the passive collagen induced arthritis using JNK2 knockout mice.30 Although some joint protection was seen, the decrease in cartilage and bone damage was modest compared with the JNK inhibitor. These data suggest that an inhibitor will need to block at least JNK1 and JNK2. It is not known if a particular alternatively spliced variant is important in FLS.
The MAP kinases are regulated by upstream kinases known as MKKs. Two of these, MKK4 and MKK7, can efficiently phosphorylate JNK on tyrosine and threonine residues, respectively, in the TXY motif.31 MKK4 can also activate p38 under some circumstances. The biological activity of JNK is markedly enhanced by dual phosphorylation, although threonine is probably the more important residue as determined by in vitro kinase assays.32 The patterns of MKK activation are dependent on the type of stimulus in many cells; cytokines primarily activate MKK7, whereas environment stresses like ultraviolet light engage MKK4.
The upstream kinases that activate JNK in RA synovium and synoviocytes have recently been examined. Western blot analysis and immunohistochemistry show that both MKK4 and MKK7 are expressed in RA, especially in the synovial intimal lining. As expected, similar levels were seen in OA synovium. Using phospho-MKK4 and -MKK7 antibodies, high levels of activated MKK4 and MKK7 were detected in RA synovial lining compared with OA. Of interest, vascular endothelial cells also expressed high levels of the phosphorylated kinases. These data suggest that JNK can be activated in situ by its immediate upstream MAPKKs in RA.
The observation that phosphorylated MKK4 and MKK7 protein expression is localised in the intimal lining layer suggests that type B synoviocytes are involved. This was confirmed in cultured synoviocytes, which constitutively express the two MAPKKs. As with synovial tissue, OA and RA derived cells had similar profiles. Both kinases were transiently activated within minutes after cytokine stimulation. The participation of MKK4 was striking in light of its relative unresponsiveness to cytokines in other cell types, including embryonic fibroblasts. Because MKK4 and MKK7 synergistically activate JNK, these data suggest that the rheumatoid cytokine milieu is an especially effective mechanism for engaging the JNK pathway in FLS (fig 3).

{kind=link}
{kind=link}
{kind=link}
Components of the JNK signalling complex. A JNK signalling complex, or signalsome, facilitates the phosphorylation of threonine and tyrosine on JNK by MKK4 and MKK7. The activated complex can then phosphorylate c-Jun on two sites and markedly increase the transcriptional activity of AP1.
As noted for the IKK pathway, various signalling proteins can organise into complexes that can include multiple enzymes in series along with scaffold proteins. Similar spatial relationships have been established for the MAP kinase cascades. The formation of complexes can theoretically enhance the efficiency as well as facilitate the engagement of specific pathways. ERK, for instance, binds specifically to a docking site on the amino terminus of MEK1 and MEK2 and is required for efficient signalling.33 Scaffold proteins like MP-1 also interact with ERK1 and MEK1 to stabilise the complexes.34
Similar MAP kinases-MAPKK complexes contribute to the formation of JNK complexes, including JNK, MKK7, and the scaffold protein JIP-1. However, the contribution of MKK4 has been uncertain until recently. Immunoprecipitation studies on the lysates of cultured synoviocytes show that MKK4 and JNK co-precipitate, as well as JNK and MKK7.35 Perhaps more interesting, MKK4 and MKK7 also co-precipitate with JNK, forming a new tripartite complex called the JNK signalsome. Confocal microscopy confirms that the three kinases co-localise in the cytoplasm, and a portion migrates to the cell nucleus after cytokine stimulation.
The functional relevance of the signalsome was demonstrated using in vitro kinase assays and GST-c-Jun as a substrate for the immunoprecipitated complex. When synoviocytes were stimulated with IL1, the MKK4 and MKK7 immunoprecipitates efficiently phosphorylated the substrate. Kinase activity was blocked if the selective JNK inhibitor SP600125 was added to the kinase reaction. Therefore, JNK is responsible for c-Jun phosphorylating function of the signalsome and the complex is activated by proinflammatory cytokines. The JNK signalsome comprised at least three kinases (JNK, MKK4, and MKK7) and may have a critical role in MAPK activation and expression of genes regulated by AP1. JNK, its upstream kinases, or interactions that maintain the structural integrity of the signalsome could be targeted as a therapeutic intervention in RA.
The situation for JNK can potentially become more complex when the top tier of the MAP kinase cascade is considered. More extended complexes with upstream MAP kinase kinase kinases (MAP3Ks) could further contribute to the amplification of cellular responses to inflammation. The MAP3K system is extraordinarily diverse and complex, and multiple members, including MEKK1, -2, -3, -4, ASK1, TAK1, and Tpl, can potentially activate JNK.36 Preliminary qualitative protein and mRNA studies were performed to determine which of the MAP3Ks are expressed in synovial tissue and cultured synoviocytes. Reverse transcriptase-polymerase chain reaction studies on OA and RA synovium demonstrate gene expression for some of these (for example, MEKK1, MEKK2, ASK1, and TAK1), but not the others. Western blot analysis and immunohistochemistry suggest that MEKK2 is highly expressed in these samples.
In vitro kinase assays show that MEKK2 kinase activity is increased by cytokines in synoviocytes and that MKK4 and MKK7 can serve as substrates. Immunoprecipitated MEKK2 complexes also contain JNK, MKK4, and MKK7, and can phosphorylate c-Jun. Therefore, an extended signalling complex containing MEKK2, MKKs, and JNK can serve as a unit to focus and amplify responses that involve AP1 components and, ultimately, metalloproteinase production. Based on the expression data, it is tempting to implicate MEKK2 as the key MAP3K. However, many other upstream kinases may potentially contribute and form additional complexes that participate.
CONCLUSIONS AND CAVEATS
Careful evaluation of the signalling networks in RA can identify key pathways and provide organisational structure. However, it is not yet possible to determine whether blockade of the targets identified by this approach will be safe and/or effective in chronic inflammatory diseases. However, the systematic dissection of signalling networks can at least contribute to the rational target selection for therapeutic interventions. Even the extraordinarily complex MAP kinase pathways can now be organised into functional groups, including signalsomes that contain only limited numbers of MAPK, MAPKK, and MAP3Ks that are likely to be relevant to arthritis. By focusing on pathways with some site and event specificity, the risk of toxicity can potentially be decreased. Examples include JNK2, which is the most relevant JNK in synoviocytes, or p38α, which is a key regulator of cytokines in macrophages compared with the other isoforms. Alternatively, disrupting signalling complexes that facilitate specific transduction systems, such as the JNK signalsome, is also possible.
One additional issue that will probably hinder this approach to kinases is the difficulty in developing (and proving) true selectivity of a small molecule inhibitor for a particular kinase. Many of these compounds bind the ATP binding site, and specificity is a significant technical hurdle. Because of the large number of kinases, it is virtually impossible to screen against all possible enzymes and ATP binding proteins with current technology. If a compound blocks more than one target, it multiplies the risk of toxicity. Hence, the twin problems of selectivity and toxicity need to be overcome to facilitate drug development in this field. Despite these reservations, the rich potential for altering the basic pathogenic mechanisms of disease suggests that it is worth the risk. Like anticytokine therapy, methodical experimentation to dissect and understand signal transduction could lead to new treatments for our patients with inflammatory and immune mediated diseases.