
To the Editor:
Majeed syndrome (OMIM #609628) is a syndromic form of chronic recurrent multifocal osteomyelitis (CRMO) that presents with early onset, severe CRMO, and a microcytic dyserythropoietic anemia of variable severity. Onset in all reported cases has been in the first 2 years of life1,2,3. A minority of patients develop a neutrophilic dermatosis. The disease is due to mutations in LPIN2, which encodes LIPIN2, a phosphatidate phosphatase important in lipid metabolism2,3,4,5,6.
Here we present the case histories of 2 male cousins of Indian heritage with Majeed syndrome. Both born to parents with consanguineous marriages, the proband presented with typical Majeed syndrome (onset of CRMO by 2 yrs of age, significant microcytic dyserythropoietic anemia, and failure to thrive), but the cousin had later onset, milder disease more reminiscent of nonsyndromic CRMO (onset at 8 yrs of age and minimal anemia). Both were found to be homozygous for the same novel LPIN2 mutation. Our report details the natural history of the disease in these 2 boys, expands the at-risk ethnic and racial group of Majeed syndrome, and demonstrates that the disease can present in older children and have a milder phenotype similar to nonsyndromic CRMO.
Case 1
A 15-year-old boy presented with a history of monthly recurrence of painful swelling of the knees, ankles, elbows, and wrists at 2 years of age lasting 10 to 15 days without rash or fever. Laboratory studies revealed chronic microcytic anemia [hemoglobin 7.9–9.9 g/dl, mean corpuscular volume (MCV) 64.6–71.4], mild leukopenia (2840–4230 cells/mm3), and elevated erythrocyte sedimentation rate (ESR; 53–140 mm/h). He had been diagnosed with juvenile idiopathic arthritis and treated with methotrexate (MTX), naproxen, and oral corticosteroids with some benefit. He was seen by the first author at 15 years of age, at which time he had severe failure to thrive (height 140 cm and weight 26 kg, both < third centiles) with absent secondary sexual characteristics. He had periarticular warmth and swelling in the lower extremities and hepatosplenomegaly but no rash. Bone marrow examination revealed marked erythroid hyperplasia with significant dyserythropoiesis (20% nuclear budding and karyorrhexis). Bone scan showed increased uptake in the distal femurs, proximal and distal tibiae, distal radii, and the left calcaneum. Magnetic resonance imaging (MRI) of the lower extremity demonstrated increased T2 signal in the proximal and distal metaphyseal regions of the bilateral tibia, suggestive of osteomyelitis. The parents did not consent to a bone biopsy.
A clinical diagnosis of Majeed syndrome was suspected. He was switched to parenteral MTX and given pamidronate 1 mg/kg/dose (except the first dose was 0.5 mg/kg) for 3 days every 3 months for over 2 years. His symptoms decreased by about 50%–60% (self-reported), and he began to grow (from 145 cm at 16 yrs to 159 cm at 19.5 yrs) and gain weight (28 kg at 16 yrs to 43 kg at 19.5 yrs), and had onset of puberty. Hemoglobin increased to 9.5 g/dl and ESR fell from 107 mm/h prior to pamidronate to ∼60 mm/h posttreatment.
Case 2
The proband’s 13-year-old cousin, also born to consanguineous parents, was asymptomatic until 8 years of age when he developed painful swelling at the metaphyseal regions of the long bones (symptoms were less severe and less frequent vs the proband). There was no fever or rash. His height and weight were between the fifth and 10th centiles. Painful juxtaarticular inflammatory swelling was present at the ankles, knees, and wrists. There was no organomegaly or rash. Investigation revealed a normal white blood cell count, mild anemia (hemoglobin 11.2 g/dl, MCV of 86.8), and a moderately elevated ESR (45 mm/h). Bone scan revealed symmetric uptake at the metaphyses of the long bones (Figure 1) and plain films showed osteolytic lesions at the fibular metaphysis bilaterally. MRI and bone marrow were not performed. He started treatment with weekly parenteral MTX with improvement in ESR (11–29 mm/h), but no significant change in hemoglobin. He reported a 70%–80% reduction in symptomatology.
{kind=link}
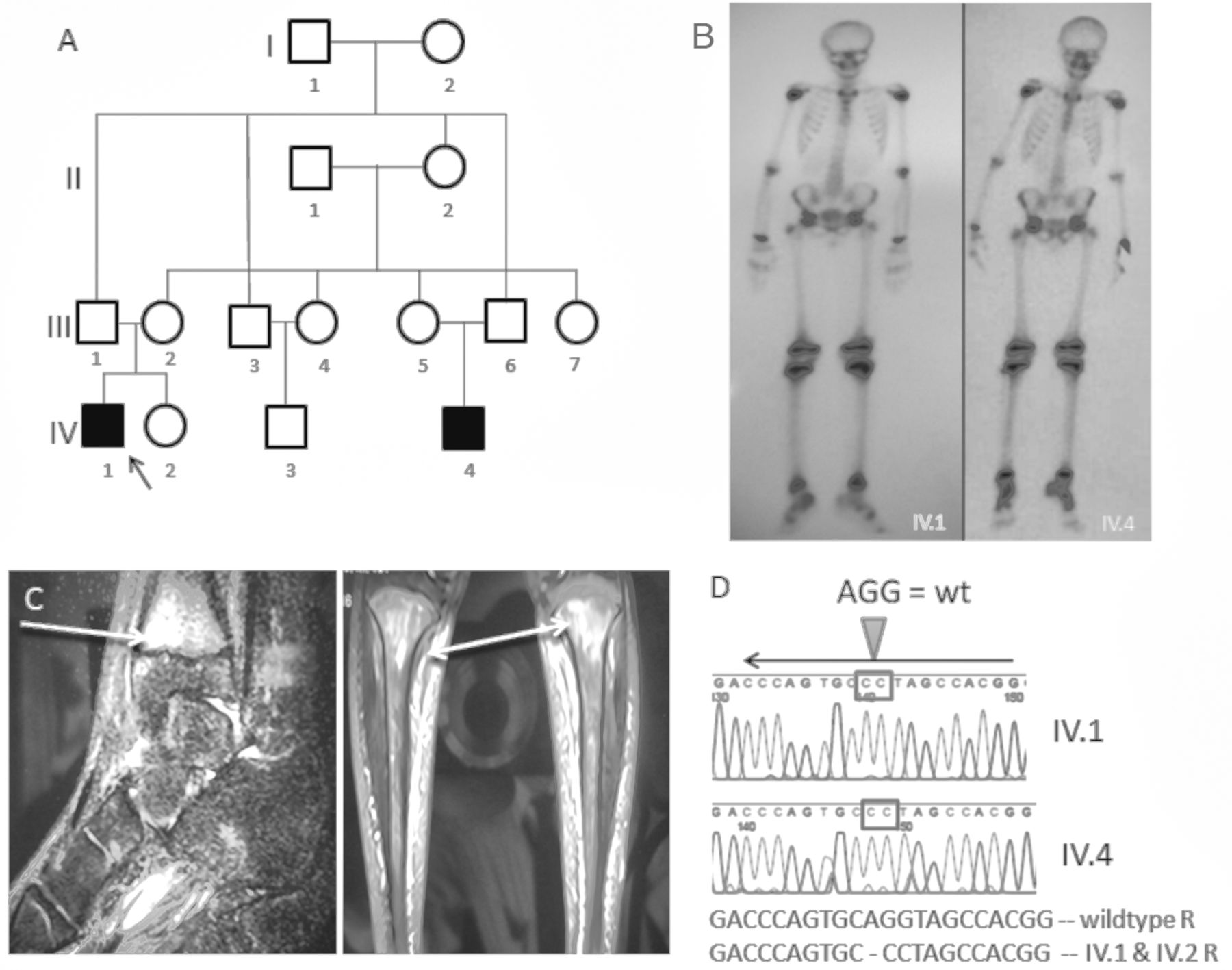
Majeed syndrome with phenotypic variability. (A) Pedigree. Affected IV1 (proband) and IV4 (cousin) are in solid black. (B) Tc99 bone scan (proband = IV1) shows increased uptake in the knees, ankles, and the right calcaneus in the proband. His cousin (IV4) shows symmetric uptake in the epiphyses of the long bones of the lower extremities. (C) Magnetic resonance imaging shows increased signal intensity in the tibial metaphyses in the proband. (D) Sequencing on IV1 and IV2 show homozygous c.2241_2243delinsGG LPIN2 mutation.
LPIN2 was sequenced as previously described4 and a homozygous deleterious mutation was identified (c.2241_2243delinsGG), which resulted in a premature truncation of the protein at amino acid 747 [p.Tyr747*] in both the proband and his affected cousin, while the parents were heterozygous.
Majeed syndrome is an exceedingly rare autoinflammatory bone disease. The disease has been described in only 9 children, all of Middle Eastern heritage1,7,8. All have had severe disease with onset in infancy to 2 years of age. Here we report, to our knowledge, the first cases of Majeed syndrome in individuals of Indian heritage and with variable severity. The proband had typical Majeed syndrome with failure to thrive, hepatosplenomegaly, delayed puberty, and dyserythropoietic anemia with onset of severe CRMO by 2 years of age. Yet, the proband’s cousin had only very mild anemia and mild CRMO with onset at 8 years of age, demonstrating for the first time that Majeed syndrome can present later in childhood with less severe disease mimicking nonsyndromic CRMO.
Interleukin 1 (IL-1) inhibitors were not available to treat these boys; however, for severe disease, IL-1β blockade has been reported to be highly effective at controlling osseous and systemic inflammation3. These cases demonstrate that there can be phenotypic variability in Majeed syndrome, and the at-risk ethnic/racial groups that can be affected by this disease can expand.
Footnotes
Supported by the US National Institute of Arthritis and Musculoskeletal and Skin Diseases at the National Institutes of Health (R01AR059703 PJFA).
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.