
Abstract
Objective. This randomized, double-blind, phase III study evaluated the efficacy and safety of ketoprofen in an ultradeformable vesicle gel compared with ketoprofen-free gel in osteoarthritis (OA) knee pain.
Methods. Patients with American College of Rheumatology-defined OA of the knee and moderate pain were randomized to receive 100 mg ketoprofen in 4.4 g transfersome gel (IDEA-033) or 4.4 g ketoprofen-free vehicle (TDT 064) topically, twice daily, for 12 weeks. The primary endpoint was mean change in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale score from baseline to Week 12.
Results. Patients (n = 555) were randomized and treated. Mean baseline WOMAC pain scores were 5.2 (SD 1.0) for IDEA-033 and 5.3 (SD 1.0) for TDT 064. Mean change in WOMAC pain scores from baseline to Week 12 was 38.6% for IDEA-033 and 44.6% for TDT 064 (Mann-Whitney estimator 0.4505; p = 0.022). Both groups reported progressive decreases in pain and improvements in function and stiffness. Mean baseline WOMAC function scores decreased from 5.4 to 3.4 with IDEA-033 and 3.1 with TDT 064 at Week 12. The proportion of patients achieving ≥ 50% decrease in WOMAC pain score from baseline at Week 12 was 41.2% (95% CI 0.35–0.47) with IDEA-033 and 50.5% (95% CI 0.45–0.57) with TDT 064. Mild skin and subcutaneous tissue disorders were the most frequently reported treatment-related adverse events (AE).
Conclusions. IDEA-033 was inferior to drug-free gel (TDT 064) in relieving moderate OA knee pain and improving joint function (Clinical Trials NCT00722852).
- OSTEOARTHRITIS
- NONSTEROIDAL ANTIINFLAMMATORY DRUGS
- PAIN
- TOPICAL
- KNEE
Osteoarthritis (OA) is a major cause of pain and disability globally, and results in significant joint pain, functional limitation, and impaired quality of life1,2,3. The prevalence of OA increases with age, therefore many patients also have other comorbid conditions, such as diabetes and cardiovascular (CV) disease, adding to the difficulties in effectively treating the condition3,4. Current pharmacologic, nonpharmacologic, and surgical treatments aim to relieve pain and joint stiffness, and to maintain or improve physical function5,6.
While widely used to ease the pain associated with OA, oral nonsteroidal antiinflammatory drugs (NSAID) have the potential to cause substantial systemic adverse events (AE), especially within the gastrointestinal (GI)7, renal8, and CV systems9,10. Oral NSAID are contraindicated, or can only be used with caution, in patients with certain comorbidities or those receiving certain concomitant medications. Topical application of NSAID reduces the potential for systemic toxicity11, but pharmacokinetic absorption is variable12.
Transfersome vesicles (IDEA AG) are ultradeformable phospholipid vesicles developed to deliver high concentrations of drugs to subdermal tissue. The excipients of the vesicles are all widely used in a variety of pharmaceutical, food, and cosmetic products. A topically applied formulation of ketoprofen in transfersome gel (IDEA-033) has been investigated in patients with OA13,14,15, having previously demonstrated efficacy against joint pain and soft tissue inflammation16. The clinical benefit afforded to patients in previous studies of IDEA-033 was controversial13,15. Thus, a new clinical development program was initiated that consisted of 2 trials, including the current study. The first study compared 2 doses of IDEA-033 and matched drug-free vehicle (TDT 064) to 200 mg celecoxib daily or oral placebo17, and the current study compared 4.4 g transfersome gel containing 100 mg ketoprofen with 4.4 g of TDT 064. Both studies investigated 12 weeks of treatment with the same design. The major difference to the previous development program was the focus on moderate pain and the use of a non-flare design.
MATERIALS AND METHODS
Study design and treatment
This was a prospective, randomized, double-blind, phase III study conducted at 39 centers across the United States (www.ClinicalTrials.gov NCT00722852). Patients receiving concomitant analgesics at screening were taken off their current medication and asked to return for their first baseline visit (B1) following a washout period of ≥ 5 days or 5 times the half-life of the analgesic. Patients with no concurrent analgesic use at screening had their B1 visit after a washout period of ≥ 5 days. For all patients, the second baseline visit (B2) took place 2–5 days after B1.
Eligible patients were randomized at B2 using a random permuted block scheme to receive 100 mg ketoprofen in 4.4 g transfersome gel (IDEA-033) or 4.4 g ketoprofen-free vehicle (TDT 064). Treatment was administered topically twice daily at 12-h intervals. A telephone evaluation of AE, rescue medication, and concomitant medication was conducted on Day 8 ± 1, with study visits scheduled at Week 2, Week 6, and Week 9. Week 12 evaluations comprised an average of part 1 of the final visit (F1) and the last day in the study (F2), which took place 2–5 days after F1.
Rescue medication (500 mg paracetamol, up to 4 times per day; total 2 g) was permitted for the treatment of intermittent pain, but not within 24 h of the next study visit or between B1 and B2. Patients who required ≥ 2 g/day of rescue or other analgesic medication for > 3 consecutive days were considered to be treatment failures and were withdrawn from the study.
Patients
Eligible patients had to be aged > 45 years with a primary diagnosis of Functional Class I–III OA of the knee according to the American College of Rheumatology clinical classification criteria. Patients had to be able to identify a predominantly painful (index) knee, and to experience moderate pain in the index knee when walking on a flat surface, defined as a score of ≥ 4 on question 1 of the Western Ontario and McMaster Universities (WOMAC) OA Index [11-point Numerical Rating Scale (NRS), version 3.1, range 0–10], and a total average WOMAC pain subscale score of < 7 (range 0–10) at B1 and B2. Female patients of child-bearing potential were to use suitable birth control methods and to have a negative pregnancy test at screening and B2.
Patients were excluded if the difference in pain rating (determined from question 1 of the WOMAC 11-point NRS) or the difference in the total average pain score between B1 and B2 were > 2 (range 0–10). Additional exclusion criteria were skin lesions or dermatologic diseases in the treatment area, extreme obesity (body mass index > 37 kg/m2), symptomatic ipsilateral hip OA, inflammatory arthritis, malignancy within the past 2 years, epilepsy, schizophrenia, or any pain condition requiring the chronic use of pain medication. Patients with known hypersensitivity or allergy to NSAID, including ketoprofen, or with preexisting asthma or bronchospasm following NSAID use were also excluded. However, patients with known GI and CV risk factors for NSAID use were eligible.
Not permitted were intraarticular injections of hyaluronic acid products in the index knee, arthroscopy of the index knee, or use of tricyclic antidepressants within 3 months prior to or during the study; use of oral, inhaled, or parenteral corticosteroids within 2 months prior to or during the study; and intraarticular injections of corticosteroids in the index knee within 1 month prior to or during the study. Receipt of any investigational product within 30 days of the screening visit, or participation in any previous clinical trial of ketoprofen in transfersome gel was also prohibited.
The study and all related documents were approved by an Institutional Review Board, and the study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from each patient prior to any study-related procedure.
Efficacy assessments
Patients completed the WOMAC pain, physical function, and joint stiffness subscales at baseline (B1 and B2), Week 2, Week 6, Week 9, and Week 12 (F1 and F2). Change from baseline (average of B1 and B2) at Week 12 (average of F1 and F2) on the pain subscale of the WOMAC (11-point NRS) was used as the primary efficacy endpoint. Patients were defined as responders if they achieved ≥ 50% decrease in WOMAC pain subscale score from baseline at Week 1218. Patients’ global assessment of response to therapy was assessed at Week 2, Week 6, and Week 9 using a 5-point categorical Likert scale ranging from 0 (none; ineffective) to 4 (excellent; virtually pain-free). Between baseline and Week 2, patients used diaries to record their pain every evening on an 11-point NRS, ranging from 0 (no pain) to 10 (pain as bad as you can imagine). Time to onset of a sustainable minimum detectable pain relief (time until ≥ 1 point improvement on the NRS sustained through Day 14) and time to onset of a sustainable meaningful pain relief (time until ≥ 2 point improvement on the NRS sustained through Day 14) were calculated from the patient diaries.
Safety
Vital signs, body weight, and temperature were recorded at screening and at each study visit, with a physical examination performed at screening and at the final visit. AE were recorded throughout the study and for 30 days after discontinuation of the study drug. The intensity of AE was rated by the investigator as mild, moderate, or severe. AE with causal relationship rated as certain, probable/likely, or possible, were considered to be drug related. Clinical laboratory assessments were performed at screening, B1, Week 6, and at the final visit.
Statistical analyses
The primary efficacy analysis was performed in the intent-to-treat (ITT) population, which included all patients who received at least 1 dose of study drug. Safety analyses were conducted in patients who received at least 1 dose of study drug and had contact with the study investigator.
The primary efficacy endpoint was the change from baseline at Week 12 on the pain subscale of the WOMAC (11-point NRS). Missing values were imputed using the last observation carried forward approach. Secondary endpoints were the change from baseline (average of B1 and B2) at Week 12 on the WOMAC stiffness and function subscales (11-point NRS); the patient global assessment of response to therapy at Week 12 (5-point Likert scale); the responder rate at Week 12; and the times to onset of both sustainable minimum detectable and meaningful pain relief during the first 2 weeks of the study.
Superiority of IDEA-033 versus TDT 064 in relation to the primary efficacy endpoint was determined using the nonparametric Mann-Whitney (MW) U test. As a benchmark for relevant baseline differences, a MW estimator of 0.36 and 0.64, respectively, were applied (referring to a standardized difference of 0.5 according to Cohen, which is regarded as a medium-sized difference)19. Stratified analyses were performed on patients receiving or not receiving analgesics at screening, with adjustment of the primary efficacy results for potential heterogeneities (Cochran-Mantel-Haenszel pooling procedure).
Sample size calculations were based on demonstrating the superiority of IDEA-033 to TDT 064 in relation to the primary efficacy endpoint, using a 1-sided superiority test with α = 0.0125, β = 0.1, and the location parameter MW statistic = 0.60. Under these assumptions, 180 patients per group were required to confirm superiority. Allowing for dropouts, 200 patients were to be randomized to each group.
We used a conservative imputation method to account for any bias introduced by the concomitant use of analgesics. If the evaluation of efficacy at the final visit was potentially biased by the intake of rescue or other analgesic medication (i.e., within 5 half-lives), a conservative approach was taken. If the observed value was better than baseline, or better than the value of the preceding visit, and the preceding value was worse than baseline, then the value from the preceding visit was used. If the preceding value was better than or equal to baseline, the replacement value was the baseline value. If the observed value was worse than the baseline value, no replacement was performed.
RESULTS
Patients
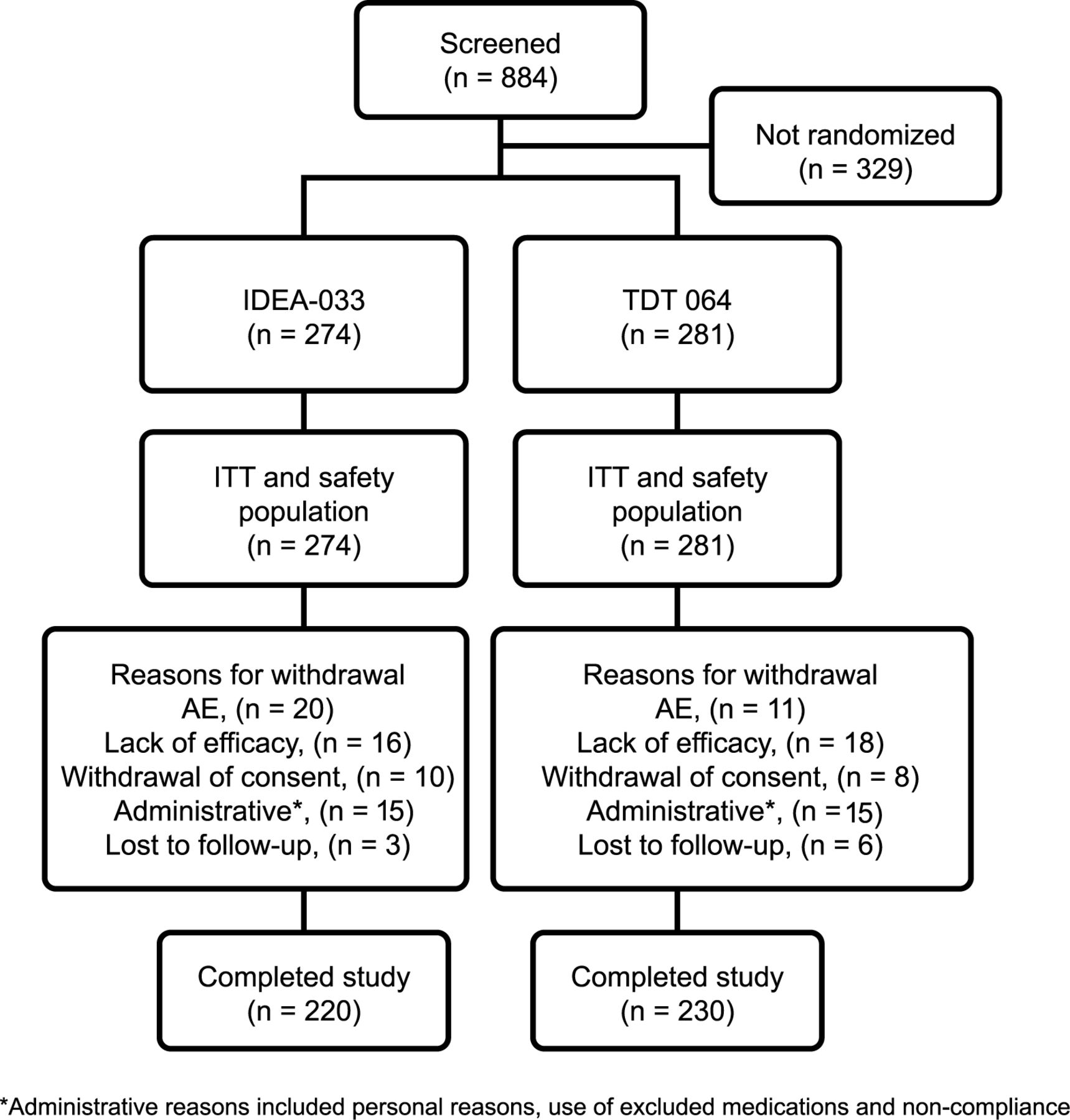
Screening occurred more rapidly than expected, and for ethical reasons all screened patients were allowed to enroll. Thus, between June 2008 and March 2009, 555 patients were randomized and received treatment (Figure 1). Patient demographic and baseline characteristics of the ITT population were balanced between the treatment groups (Table 1). Mean WOMAC pain subscale scores at baseline were 5.2 (SD 1.0) in the IDEA-033 group and 5.3 (SD 1.0) in the TDT 064 group. No statistically relevant group differences were observed.
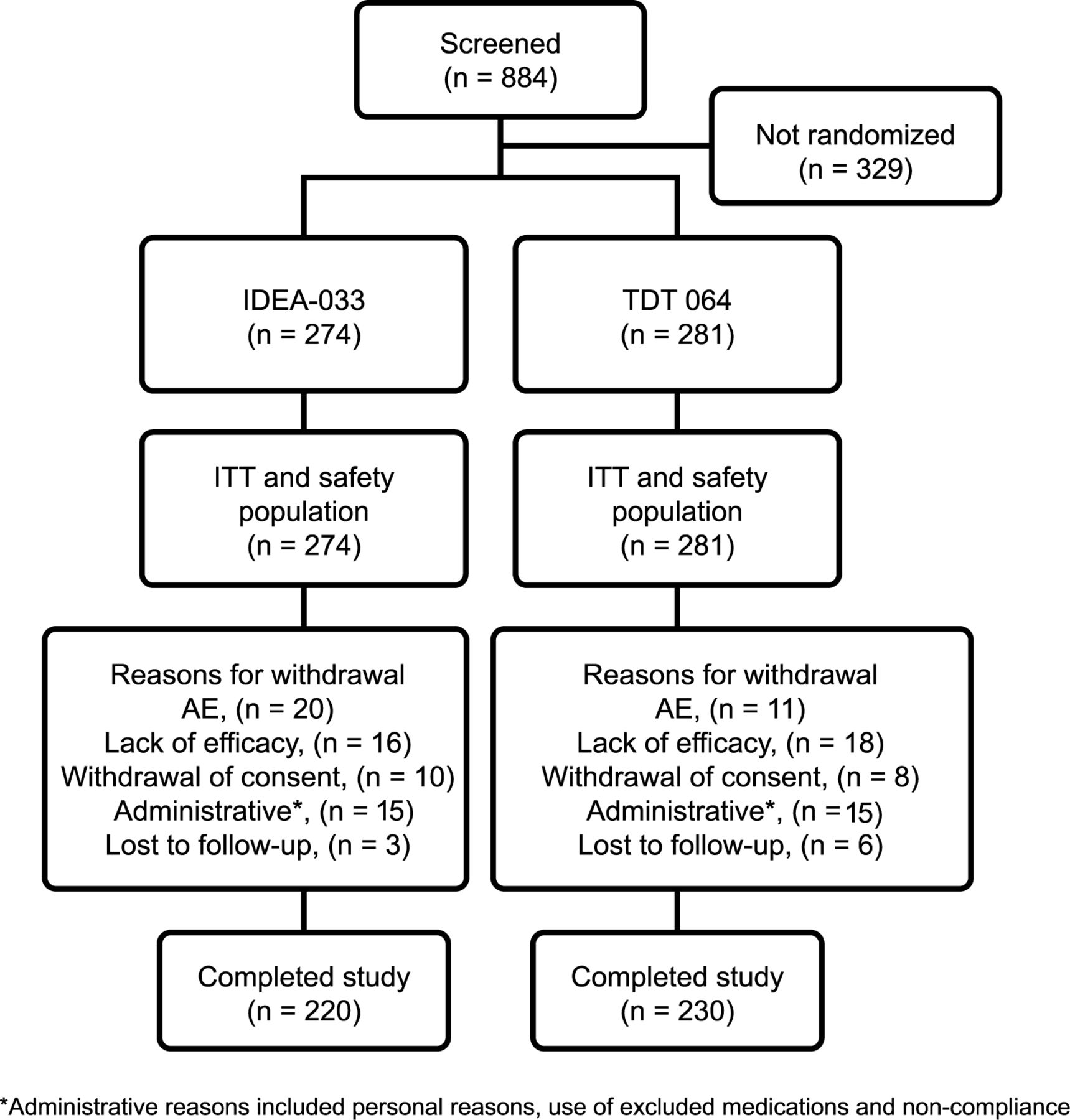
Patient disposition and flow through the study. ITT: intent-to-treat; AE: adverse event.
Patient demographic and baseline characteristics [intent-to-treat (ITT) population].
Efficacy
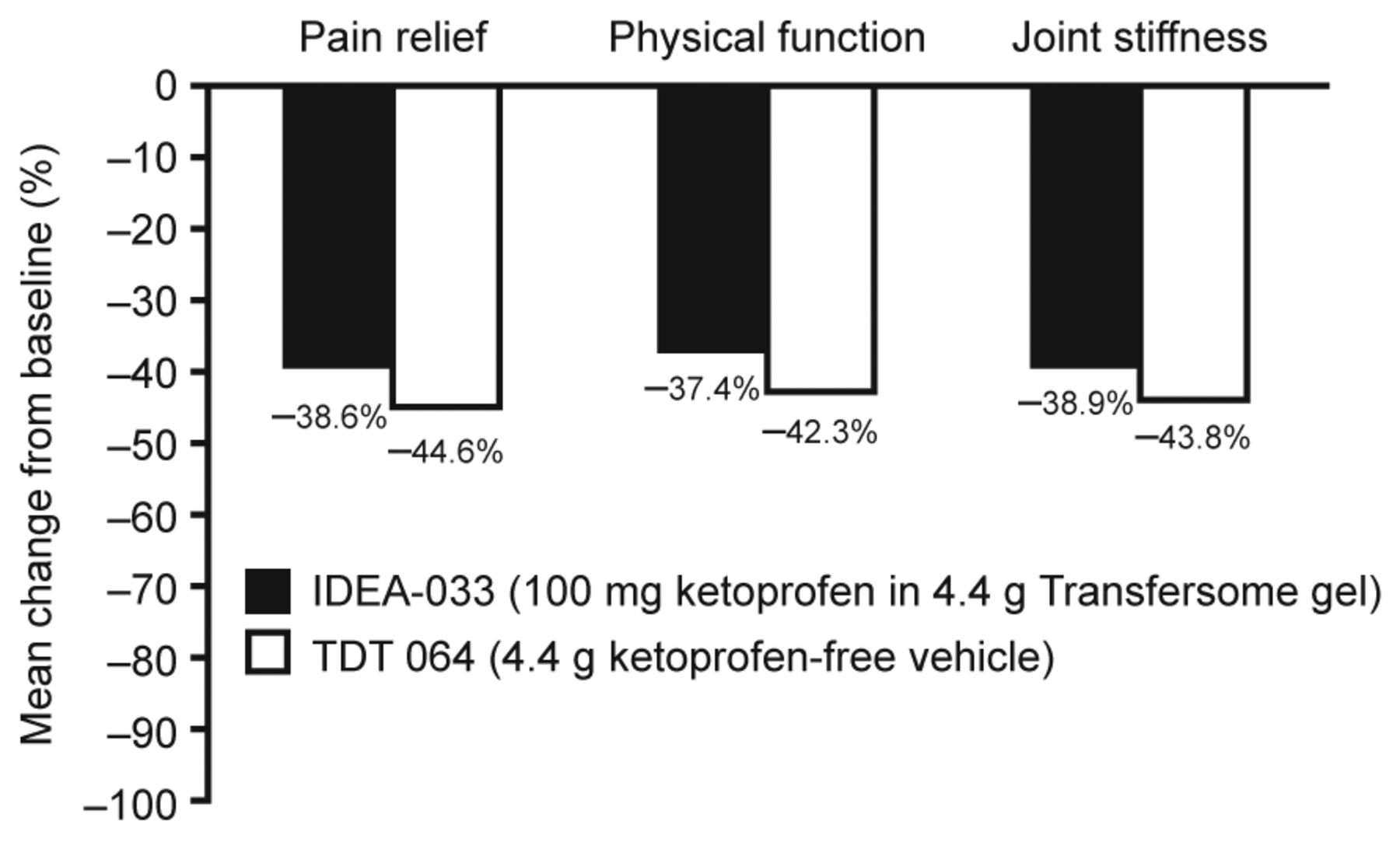
The mean percentage change in WOMAC pain subscale scores from baseline to Week 12 was 38.6% (SD 37.9) for IDEA-033, as compared with 44.6% (SD 39.0) for TDT 064 (MW estimator 0.4505; p = 0.022 in favor of TDT 064; Figure 2). Both treatment groups reported progressive pain decreases throughout the study (Table 2). Week 12 mean WOMAC pain subscale scores were 3.2 (SD 2.1) in the IDEA-033 group and 2.9 (SD 2.2) in the TDT 064 group.

Mean percentage change from baseline to Week 12 in WOMAC pain subscale, physical function, and joint stiffness scores (ITT population). WOMAC: Western Ontario and McMaster Universities Osteoarthritis Index; ITT: intent-to-treat.
Mean change from baseline in WOMAC pain subscale, physical function, and joint stiffness scores (ITT population).
Improvements in physical function and joint stiffness were also progressive and comparable in both treatment groups throughout the study (Table 2). Mean WOMAC function subscale scores decreased from 5.4 (SD 1.2) in both the IDEA-033 and TDT 064 groups at baseline to 3.4 (SD 2.2) and 3.1 (SD 2.2), respectively, at the final visit. Similarly, mean WOMAC stiffness subscale scores decreased from a baseline value of 5.8 (SD 1.4) and 5.7 (SD 1.4) in the IDEA-033 and TDT 064 groups, respectively, to final values of 3.6 (SD 2.3) and 3.2 (SD 2.3), respectively. IDEA-033 was statistically inferior to TDT 064 in the mean percentage change from baseline in WOMAC function [37.4% (SD 36.4) vs 42.3% (SD 39.4), respectively; MW estimator 0.4570; p = 0.04 in favor of TDT 064] and stiffness subscale scores [38.9% (SD 37.4) vs 43.8% (SD 38.2), respectively; MW estimator 0.4655; p = 0.08 in favor of TDT 064; Figure 2].
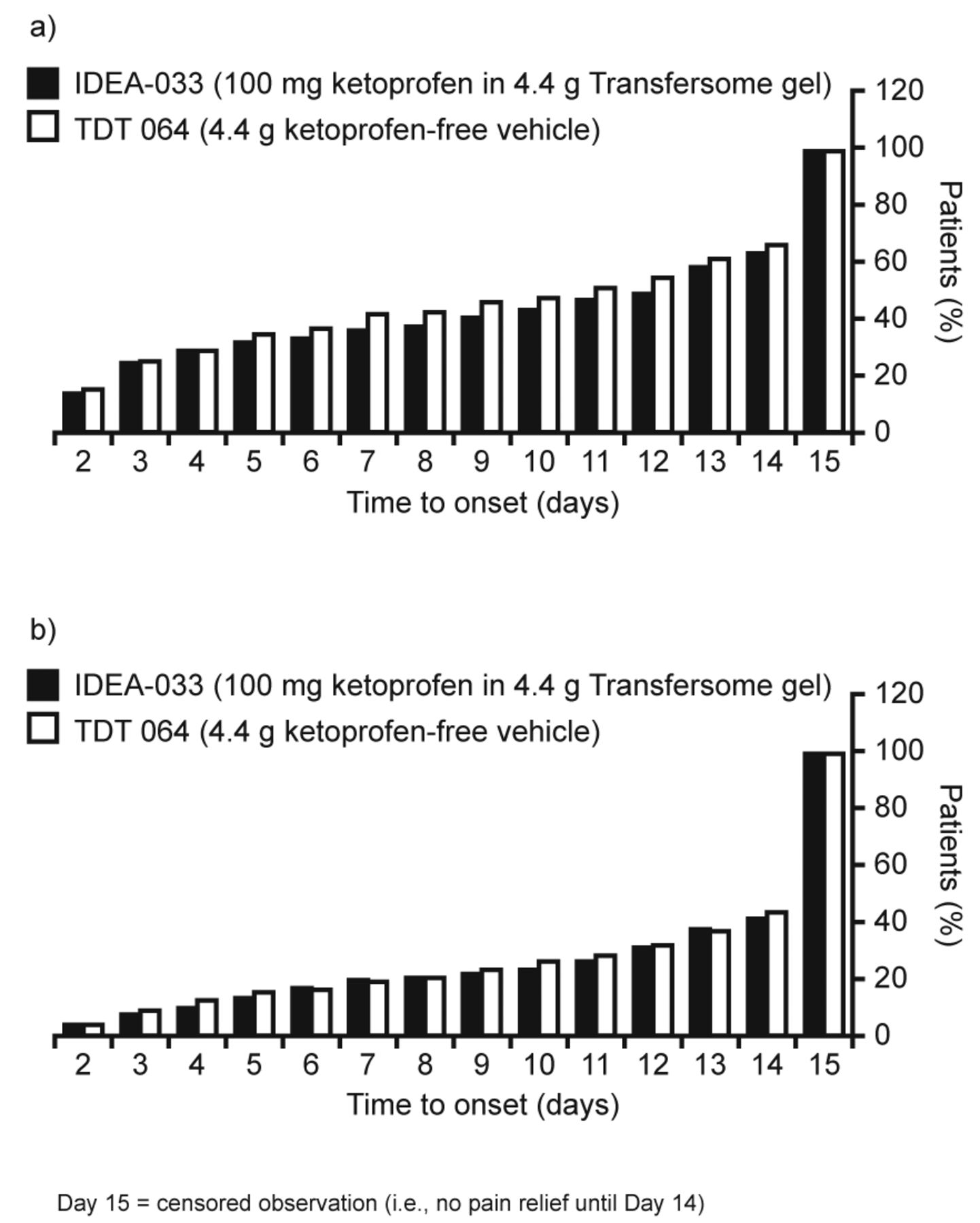
Analysis of the time to onset of a sustainable minimum detectable pain relief revealed a similar time to pain relief in the TDT 064 group compared with the IDEA-033 group (Figure 3a). Pain relief was reported at 2 days from the start of the study (earliest timepoint measured) in 16.0% of patients receiving TDT 064 and in 14.6% of patients receiving IDEA-033. Time to onset of sustainable meaningful pain relief, defined as ≥ 2 point improvement on the NRS sustained through Day 14, was similar between the treatment groups (Figure 3b).
{kind=link}
{kind=link}
{kind=link}
Time to onset of (a) a sustainable minimum detectable pain relief, and (b) a sustainable meaningful pain relief intent-to-treat population.
The responder rate at Week 12, defined as the proportion of patients achieving ≥ 50% decrease in WOMAC pain subscale score from baseline, was 41.2% (95% CI 0.35–0.47) with IDEA-033 and 50.5% (95% CI 0.45–0.57) with TDT 064. At the final study visit, response to therapy was rated as “excellent” or “good” by 54.7% of patients in the IDEA-033 group, and by 60.5% of patients in the TDT 064 group.
A substantial number of patients used analgesics, in particular paracetamol, as rescue medication in addition to the study medication (82.1% in the IDEA-033 group and 80.8% in the TDT 064 group). Because a specific imputation method was used to account for any bias introduced by concomitant use of analgesics, a sensitivity analysis was performed to compare statistical results obtained with or without correction for use of analgesics. No differences in the main outcome criteria were observed. Results from the analysis that accounted for analgesic use are reported.
Safety
In total, 221 patients (39.8%) experienced 456 AE. A similar proportion of patients in each treatment group experienced at least 1 AE: 108 patients (39.4%) randomized to IDEA-033, and 113 patients (40.2%) randomized to TDT 064. Serious AE occurred in 3 patients receiving IDEA-033 and in 4 patients receiving TDT 064; 1 of the serious events in the IDEA-033 group (headache) was assessed by the investigator as possibly related to treatment. One patient receiving IDEA-033 committed suicide 72 days after baseline; this event was classed as unrelated to study treatment. Overall, 32 patients (5.8%) discontinued the study because of AE, or a combination of AE and lack of efficacy. That number includes 20 patients receiving IDEA-033 and 12 patients receiving TDT 064. In both treatment groups, more than 50% of AE were of mild intensity.
Skin and subcutaneous tissue disorders were the most common treatment-related AE in both treatment groups, with rash [13 (4.7%) IDEA-033 patients, 8 (2.8%) TDT 064 patients] and localized erythema [9 (3.3%) IDEA-033 patients, 4 (1.4%) TDT 064 patients] reported most frequently (Table 3). Treatment-related GI disorder AE were minimal (< 1% of patients in either treatment group), and consisted of flatulence and nausea in the TDT 064 group [1 (0.4%) patient each], and hemorrhoids and upset stomach in the IDEA-033 group [1 patient each (0.4%)]. There were no reports of any treatment-related cardiac or vascular disorder AE throughout the study.
Treatment-related adverse events (AE) occurring in > 1% of patients in either treatment group (safety population).
No clinically relevant changes in physical measurements or vital signs were reported. However, a change in hematocrit levels was noted in 6 patients in the IDEA-033 group, from “not clinically significant” at baseline, to “potentially clinically significant” at Week 12 (Bowker test for symmetry < 0.05 2-sided).
DISCUSSION
The efficacy of topically applied 100 mg ketoprofen gel was not superior to matched TDT 064, and was actually statistically inferior in relieving OA knee pain in our study. Improvements in physical function and joint stiffness throughout the 12-week study were also no better with IDEA-033 versus TDT 064. Although not directly comparable because of differences in control groups, these findings are in line with results from a parallel phase III study (n = 1399) using a similar design that also showed a benefit of TDT 064 in patients with OA knee pain17. That study included both a positive control arm (oral celecoxib) and a negative control arm (oral placebo) and reported that TDT 064 was statistically superior to oral placebo and not inferior to oral celecoxib in reducing knee pain after 12 weeks of treatment. The mean reduction in WOMAC pain score at Week 12 was −1.9 (−40.8%) for ketoprofen 50 mg, −1.9 (−40.9%) for ketoprofen 100 mg, −1.9 (−39.8%) for 2.2 g TDT 064, −1.8 (−37.8%) for 4.4 g TDT 064, −1.9 (−40.4%) for celecoxib, and −1.4 (−29.3%) for oral placebo.
Response to IDEA-033 in our current study, with a 38.6% mean reduction in WOMAC pain scores from baseline, was comparable with data from other previous 12-week studies of this formulation. Improvements of 40% and 57% from baseline WOMAC pain scores were reported in 2 randomized, double-blind, phase III studies following 12 weeks’ treatment with 50 mg or 100 mg ketoprofen gel in patients with OA of the knee14,15.
The TDT 064 drug-free transfersome vesicle formulation (Sequessome, Pro Bono Bio Entrepreneur Ltd.), has attracted recent interest following the reported effects of the vehicle without an active pharmaceutical ingredient in patients with OA of the knee. In a placebo- and active-controlled, phase II study, 6 weeks’ treatment with IDEA-033 and TDT 064 both provided pain relief13. Further, in a phase III, dose-finding study (n = 867), 50 mg and 100 mg ketoprofen gel and TDT 064 were associated with marked clinical improvements in pain over a 12-week period14.
The 44.6% mean reduction in WOMAC pain scores seen with TDT 064 in our study, and the 42.3% mean improvement in WOMAC physical function scores, concur with published data reporting considerable changes from baseline with TDT 064. In a randomized, double-blind, phase III study, a 49.5% mean reduction in WOMAC pain subscale score was observed from baseline to Week 12 in patients receiving TDT 064, with a 36.1% mean improvement in WOMAC physical function scores14. Limitations of these findings include the lack of an active established control within the study and the potential influence of rescue medication on response to treatment.
A large metaanalysis of 198 randomized trials conducted by Zhang, et al concluded that the placebo effect exists in OA trials, particularly for the subjective outcomes of pain, physical function, and joint stiffness20. A number of factors were shown to significantly increase the size of the placebo effect, including the strength (effect size) of the active treatment, the patients’ disease severity at baseline, the route of delivery, and the sample size20. Topical treatments tended to elicit a greater placebo effect than oral treatments, though the difference was not statistically significant. Placebo response rates in other trials of topical agents for OA are somewhat lower than data reported for TDT 064 in our study, and in previous studies of this formulation13,14. For example, in a randomized study of 12 weeks’ treatment with topical diclofenac versus vehicle control in patients with primary OA of the knee (n = 326), a 33% mean change from baseline to Week 12 in WOMAC pain scores was noted in the vehicle control group, along with a 24% mean change from baseline in each of the WOMAC physical function and joint stiffness scores21. In a further randomized, double-blind study of 4 weeks’ treatment with topical diclofenac versus vehicle control for knee OA (n = 248), mean WOMAC pain scores in the vehicle control group changed by 26.9% from baseline, mean WOMAC physical function scores by 18.7%, and mean WOMAC joint stiffness scores by 20.0%22. This is further supported by a recent metaanalysis of the 4 randomized, double-blind, phase III trials in patients with OA of the knee (n = 1061) in which TDT 064 was administered. The effect size for TDT 064 was 1.15 (95% CI 1.09–1.21)23 compared with an effect size of 0.63 (95% CI 0.47–0.80) reported for topical placebo applications in the metaanalysis by Zhang, et al20.
The tolerability of IDEA-033 was comparable with matched TDT 064 in this patient group, including enrolled patients with known CV and GI risk factors for NSAID use. The most frequently reported treatment-related AE were skin and subcutaneous tissue disorders. Treatment-related GI disorders were minimal, and treatment-related cardiac and vascular disorders were absent. These findings add to available published safety data that highlight a lack of safety concerns with these transfersome formulations in patients with OA13,14.
Ketoprofen gel (100 mg) was inferior to TDT 064 in relieving mild-to-moderate pain associated with knee OA, and was associated with a higher frequency of withdrawals due to AE. Because of the small magnitude of difference, the superiority of TDT 064 might present a chance effect. A study with the same design supports this, showing comparable efficacy for IDEA-033 and TDT 064 at the level of 200 mg celecoxib17.
Acknowledgment
The authors acknowledge the study investigators of the IDEA-033 Study Group and the patients who participated in the study. Editorial assistance with the preparation of the manuscript was provided by Bollin Strategies Ltd., UK, and was funded by Pro Bono Bio Entrepreneur Ltd., UK.
Footnotes
-
Supported by IDEA AG, Germany (sponsor of the clinical trial). Dr. Rother was an employee of IDEA AG at the time the study was conducted and is a paid consultant of Pro Bono Bio Entrepreneur Ltd. Dr. Conaghan has participated in speaker meetings or advisory boards for Janssen, Merck, Pfizer, Pro Bono Bio Entrepreneur Ltd, and Servier.
- Accepted for publication June 28, 2013.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.