
To the Editor:
Sjögren’s syndrome (SS) is a chronic autoimmune disease characterized early in its course by lymphocytic infiltration in the salivary and lacrimal glands, resulting in the major manifestations of keratoconjunctivitis sicca and xerostomia. Presumably through a multistep lymphoproliferative process, SS has been associated with the development of non-Hodgkin’s lymphoma (NHL) in 5%–10% of patients1. Mucosa-associated lymphoid tissue (MALT) lymphomas constitute the majority of NHL subtypes seen in SS, followed by nodal marginal zone lymphomas and diffuse large B-cell lymphomas (DLBCL)2. We describe what could be the first case of primary DLBCL of the kidney, a rare extranodal lymphoma, as a lymphoproliferative complication of SS.
A 64-year-old woman presented at age 39 with polyarthralgias and morning stiffness attributed to rheumatoid arthritis (RA). She was treated with hydroxychloroquine and nonsteroidal agents. She subsequently developed dry eyes, dry mouth, difficulty swallowing without drinking fluids, and vaginal and nasal dryness, which she treated with topical moisturizing agents. At age 58, she was noted to have mild pancytopenia with leukocytes 3600/mm3, hemoglobin 11.3 g/dl, and platelets 95,000/mm3. Bone marrow examination revealed erythroid hyperplasia, megakaryocytic hyperplasia, and atypical lymphocytosis. Urinalysis showed 20–30 white blood cells/high-powered field (HPF), without red cells, bacteria, nitrite, or proteinuria.
Serological testing revealed antinuclear antibody (ANA) 1:640 speckled pattern; absence of antibodies to dsDNA and Sm/RNP; anti-SSA antibody 413 U (strongly positive); anti-SSB 129 U (weakly positive); rheumatoid factor (RF) 32 IU (normal < 16); anticitrullinated protein antibodies negative; and normal serum complement C3 and C4 levels. Serum immunoelectrophoresis revealed polyclonal elevations of IgG and IgA. Cryoglobulins, human immunodeficiency virus, and hepatitis B and C serologies were negative.
Schirmer’s test was positive (< 1 mm in 5 min). A labial minor salivary gland biopsy showing marked focal lymphocytic sialadenitis, with a focus score of 4 (> 50 lymphocytes per 4 mm2 tissue sample), was indicative of SS.
With clinical, serological, and pathologic features of SS and the absence of radiographic changes of RA, the patient’s diagnosis was revised to primary SS with active synovitis and she was treated with hydroxychloroquine, oral pilocarpine, and omega-3 fatty acid supplementation, with improvement in her sicca symptoms.
Five years later, she presented with complaints of feverishness, night sweats, anorexia, and a 40-lb weight loss over 3 months.
General physical examination was notable for the absence of palpable sialadenopathy or lymphadenopathy; dry oral mucosa, with evident dental caries; and mild conjunctival injection.
Laboratory evaluation revealed leukocytes 2400/mm3, reflecting both neutropenia and lymphopenia; hemoglobin 8.3 g/dl; platelets 59,000/mm3; ANA 1:1280 homogeneous and speckled patterns; anti-SSA antibody > 800 U; anti-SSB 189 U; RF 188 IU, type III cryoglobulins identified; absences of anticardiolipin antibodies and lupus anticoagulant; C3 60 mg/dl (normal 75–140), C4 < 8 mg/dl (9–36); persistent polyclonal elevations of IgG and IgA; IgG subclasses revealed normal distribution; urinalysis showed 59 red blood cells/HPF, without casts or proteinuria.
Repeat bone marrow aspiration showed only scattered plasma cells and few atypical lymphocytes, without other morphological abnormalities.
Computed tomography of the chest and abdomen revealed a large left renal mass extending from diaphragm to iliac crest, mild splenomegaly, small bilateral pleural effusions, mild ascites, and a small, benign-appearing, pleural-based nodule in the left lower lobe, without mediastinal, hilar, or retroperitoneal lymphadenopathy.
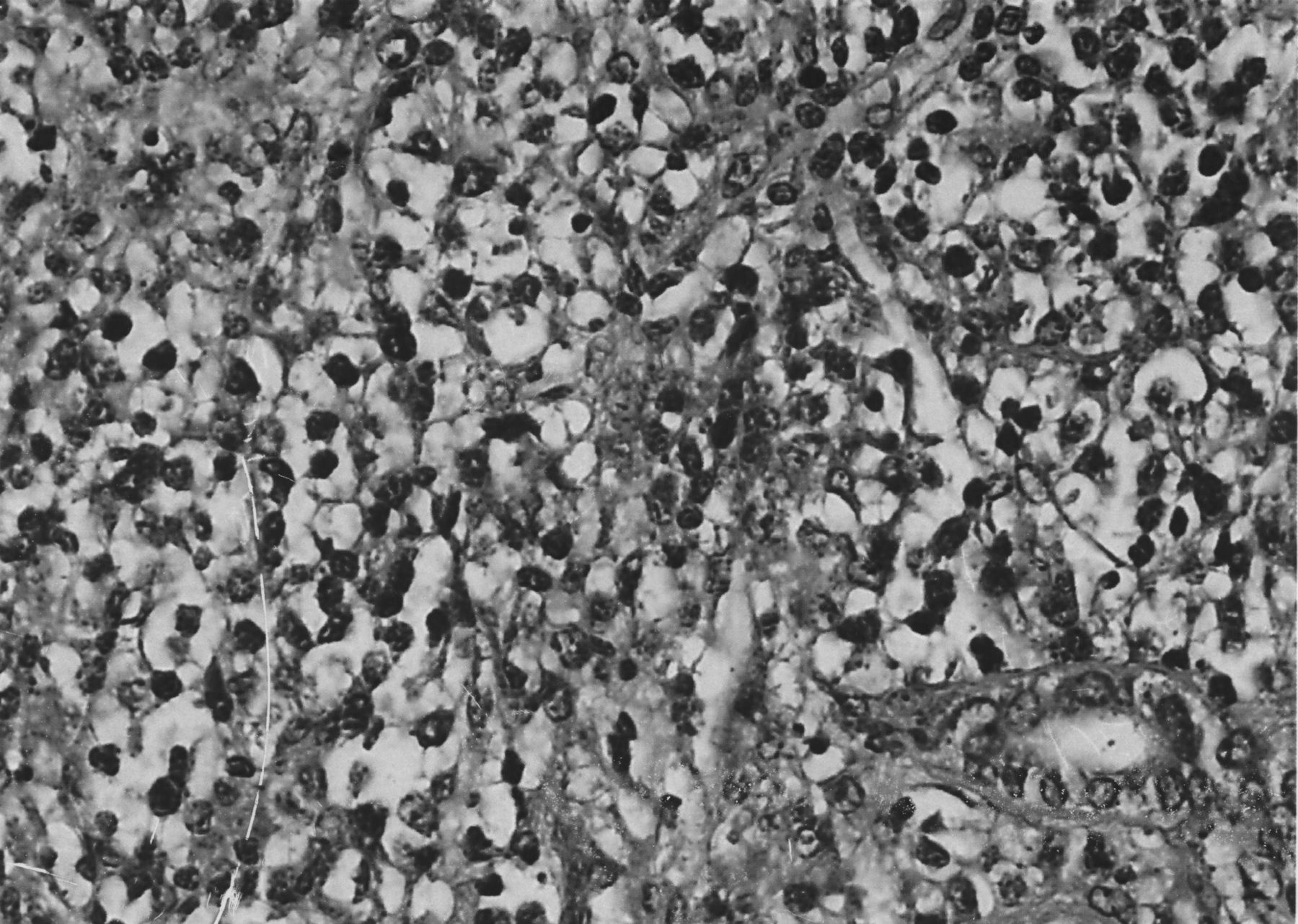
Because of suspicion of hypernephroma, left nephrectomy was performed. Histological examination revealed diffuse large-cell NHL, with immunohistochemical stains positive for leukocyte common antigen (CD45) and L26 (CD20), consistent with a B cell origin (Figure 1). Flow cytometry detected monoclonal B cells, with lambda light-chain restriction. Southern blot analysis showed monoclonal gene rearrangement of the immunoglobulin heavy chain. Bone marrow biopsy demonstrated active tri-lineage hematopoiesis, without evidence of lymphoma. Bone scintigraphy revealed increased uptake only at the site of bone marrow biopsy.

{kind=link}
Sections of the kidney show several large foci of neoplastic infiltration effacing the normal renal architecture. The infiltrating cells are large and have roundish nuclei, clumped chromatin, and obvious nucleoli. There is single-cell necrosis and many mitoses (H&E stain, original magnification 40×). In conjunction with the immunohistology, the features are diagnostic of diffuse large B-cell lymphoma.
She was treated with 6 monthly courses of cyclophosphamide, doxorubicin, vincristine, and prednisone. The dose of vincristine was reduced toward the end of the treatment because of neuropathic symptoms. Rituximab infusions were not administered. She remained disease-free until her death from valvular heart disease (with no echocardiographic evidence of thrombotic endocarditis), congestive heart failure, and renal failure, considered unrelated to lymphoma progression, 2 years later.
The relationship between SS and lymphoma has been known since the 1960s3, with recent metaanalyses supporting a relative risk of lymphoma development ranging from 5.4 to 161,4,5,6. The majority of lymphomas developing in patients with SS are low-grade marginal zone B-cell lymphomas, particularly of the MALT variety7, with major sites including the salivary glands, buccal mucosa, lacrimal glands, lung, stomach, and cervical lymph nodes2. But lymphomas in SS can also progress toward a less differentiated cell type, with DLBCL accounting for 15% of NHL cases in SS1,4,6.
Renal involvement from systemic lymphoma is not at all infrequent. However, primary renal involvement is rare, representing < 5% of NHL8. There is a reported case of a 62-year-old woman with SS who developed MALT lymphoma of the kidney, presenting with simultaneous parotid gland involvement9. MALT lymphoma of the kidney has also been reported complicating systemic lupus erythematosus10.
Because low-grade MALT lymphomas of the kidney often have an indolent clinical course with a propensity to be localized at diagnosis and curable with local therapy, it is important to distinguish MALT lymphoma from other renal lymphomas that take a more aggressive clinical course. In contrast to MALT lymphoma, DLBCL is typically characterized by aggressive histopathology, early extrarenal infiltration, and poor prognosis, with mean survival of generally < 1 year from diagnosis8.
Renal involvement with DLBCL has been reported in a 58-year-old man with Mikulicz syndrome and IgG4 lymphoproliferation, without SS, who had simultaneous involvement of stomach, ileum, pancreas, and lung nodules11.
Studies have indicated that the risk of development of lymphoma in patients with SS is linked with the presence of neutropenia, cryoglobulinemia, splenomegaly, lymphadenopathy, vasculitis, and low C4 levels1,12. The presence of any of these risk factors should justify closer scrutiny of these patients. Interestingly, patients presenting with lymphocytopenia, as did our patient, are more likely to develop the more aggressive DLBCL12. Because of the presumptive diagnosis of renal cell carcinoma, the diagnosis of DLBCL only became apparent at nephrectomy. Had there been stronger suspicion of the possibility of SS-related renal lymphoma, fine-needle aspiration biopsy might have yielded the diagnosis, averting the need for nephrectomy.
To our knowledge, the patient described here represents the first reported case of SS presenting with the less common histology, DLBCL, exclusively involving the kidney. It remains unclear how NHL develops in organs that do not typically contain lymphoid tissue. We hypothesize that the patient may have had subclinical interstitial nephritis, a recognized complication of SS13, presenting an underlying clinically silent inflammatory process that underwent malignant lymphoproliferation to DLBCL.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.