
Abstract
Objective. To analyze clinical characteristics, survival, causes of death, and risk factors associated with mortality in patients with adult-onset idiopathic inflammatory myopathies (IIM) in Japan.
Methods. We retrospectively investigated 197 patients diagnosed with adult-onset IIM at our hospital from 1984 to 2009 according to Bohan and Peter criteria for polymyositis (PM)/dermatomyositis (DM) and modified Sontheimer’s criteria for clinically amyopathic DM (ADM).
Results. Survival in the whole group at 1, 5, and 10 years was 85%, 75%, and 67%, respectively. Mortality in cancer-associated myositis was the worst (25% at 5 yrs), followed by clinically ADM (61% at 5 yrs) and primary DM (77% at 5 yrs). Primary DM had significantly low survival compared to primary PM (91% at 5 yrs; p = 0.0427). Among the 53 patients who died were 6 patients with ADM (11%) and 20 patients with primary DM (38%). Interstitial lung disease (ILD) was the main cause of death in clinically ADM (71%) and primary DM (60%), most of which occurred within the first few months. Fewer patients died in primary PM (9%) and overlap myositis (13%). Independent risk factors for death were older age (HR 1.031; 95% CI 1.009–1.053) and skin ulcers (HR 3.018; 95% CI 1.340–6.796) in the whole group and ILD with mild serum creatine kinase levels (< 500 IU/l; HR 3.537; 95% CI 1.260–9.928) in primary DM.
Conclusion. Survival of clinically ADM and primary DM was low, mainly due to fatal ILD, compared to primary PM. Establishing therapeutic strategy for ILD may improve the survival in our patient population.
- INFLAMMATORY MYOPATHIES
- POLYMYOSITIS
- DERMATOMYOSITIS
- AMYOPATHIC DERMATOMYOSITIS
- SURVIVAL
- RISK FACTORS
The idiopathic inflammatory myopathies (IIM) are autoimmune systemic diseases of unknown etiology characterized by acute, subacute, or chronic skeletal muscle inflammation. They frequently involve internal organs, mainly the pulmonary, cardiac, and gastrointestinal systems, which occasionally have considerable effect on the morbidity and mortality of IIM1. Before the generalized use of glucocorticoid, the mortality rate was reported to be as low as 52%–65% at 5 years in the most frequent myopathies — polymyositis (PM) and dermatomyositis (DM)2,3. Because an early diagnosis and aggressive use of immunosuppressive agents have become standard in recent decades, the survival of patients with IIM has improved progressively worldwide4,5,6,7,8,9,10,11.
Studies on survival of IIM, with particular interest in interstitial lung disease (ILD), have been reported from Asian countries12,13. But compared to Europe and the United States, much less evidence has been accumulated regarding longterm survival, prognostic factors, and risk factors associated with mortality in patients with IIM in our patient population. Further, because the modern definition of IIM formulated by Bohan and Peter required the presence of muscle involvement by definition14,15, amyopathic dermatomyositis (ADM), which shares typical cutaneous manifestations of DM without clinically evident proximal muscle inflammation16,17, was not included in the modern published case series of IIM18.
Our aim was to illustrate differences of clinical characteristics, longterm survival, causes of death, and associated risk of mortality of adult-onset IIM, particularly focusing on primary PM, primary DM, and clinically ADM. This is one of the largest studies of survival of patients with IIM conducted in a single institute.
MATERIALS AND METHODS
Patient selection and data analysis
We retrospectively investigated the medical record from May 1984 through October 2009 and identified 243 consecutive patients with IIM. All patients were seen by clinical staff at the Division of Rheumatology and Allergology, Department of Internal Medicine, St. Marianna University, School of Medicine. We extracted the following data: background of the patients, major systemic and organ manifestations, laboratory data at the initial presentation, causes of death, and date of death or termination of followup. Out of the 243 patients, we excluded 13 because of insufficient clinical data and 32 because initial treatment was received in other hospitals. One patient with inclusion body myositis was also excluded from this study. After the exclusion of these patients, a total of 197 patients with adult-onset IIM were analyzed. All patients were Japanese except 1 from South Asia. The protocol was approved by the ethical review board in our institute.
Diagnosis
The diagnosis of IIM was based on the criteria defined by Bohan and Peter14,15: (1) symmetrical weakness of proximal muscles; (2) elevation of muscle enzymes — creatine kinase (CK) and lactate dehydrogenase; (3) typical electromyographic (EMG) findings; (4) typical pathological changes in muscle biopsy; and (5) characteristic dermatological features. The patients who fulfilled the criteria for the diagnosis of definite, probable, and possible PM/DM were included in the study. The patients were also subdivided according to Bohan and Peter classification14,15 into the following groups: adult primary PM; adult primary DM; cancer-associated myositis (CAM); and myositis associated with other autoimmune diseases, or overlap myositis (OM). In our study, we considered a patient as having CAM when the diagnosis of cancer was made within the first 3 years after PM/DM appeared, in accordance with a previous study19. The diagnoses of rheumatoid arthritis (RA), systemic sclerosis (SSc), systemic lupus erythematosus (SLE), and Sjögren’s syndrome (SS) were based on American College of Rheumatology criteria for RA20, SSc21, and SLE22, and the European criteria for SS23.
Clinically ADM, which is the combination of ADM and hypomyopathic DM (HDM), was diagnosed by the modified Sontheimer’s definitions16,17,18,24. ADM is manifested with Gottron’s papules or heliotrope rash with no symptom or signs of muscle weakness, and normal serum CK levels, EMG, and muscle biopsy. HDM is manifested with Gottron’s papules or heliotrope rash, with no symptom of muscle weakness, and normal or mildly reduced muscle strength compatible with age, sex, and severity of systemic illness. The serum CK levels were < 1.5 times the upper normal limits. EMG was normal or with only suspicious myopathic change. The pathological findings were normal or with scant lymphocyte infiltration with normal muscle structure. Patients who did not have an EMG or muscle biopsy were treated as possible clinically ADM when they fulfilled other definitions of ADM/HDM24. Patients with premyopathic DM18 in whom fatal ILD developed within the 6 months of their disease course were also included under clinically ADM.
Clinical evaluation
Degree of muscle weakness was scored by manual muscle testing, which was based on the evaluation of 18 proximal muscle groups as described25, comprising the right and left deltoid, biceps brachii, brachioradialis, triceps brachii, iliopsoas, gluteus maximus, quadriceps femoris, and hamstring muscles, and the neck flexors and extensors. The responses were rated according to the Medical Research Council (MRC) scale (0 = lowest score and 5 = highest score), with a maximum MRC score of 90. The normal range of CK (40–200 IU/l in men, 20–160 IU/l in women) was not changed during the study period. All the patients received an initial prospective standardized evaluation of organ involvement, which resulted in the detection of systemic complications such as Raynaud’s phenomenon, dysphagia, and cardiac and pulmonary abnormality. ILD was diagnosed by chest radiograph and high-resolution computed tomography of the lungs. Myocarditis was defined on the basis of new onset of abnormal electrocardiography findings, elevation of serum troponin T, and abnormal multiple patchy defects on the thallium-scintigraphy scan after ruling out coronary artery disease. Antinuclear antibodies (ANA) and anti-Jo-1 antibodies were detected by immunofluorescence and ELISA, respectively.
Treatment
Prednisolone was administered at a dose between 0.5 mg/kg and 1 mg/kg. Methylprednisolone pulse therapy was added when the patients had severe myositis or fatal ILD. Immunosuppressants such as cyclophosphamide (28 patients), cyclosporine (27), azathioprine (22), methotrexate (4), or tacrolimus (3) were used for patients who had fatal ILD, myocarditis, or steroid-resistant myositis, but the choice of the treatment was basically left to the individual physician’s decision.
Followup periods and causes of death
Followup period was defined from initial presentation to either the date of death or the latest visit to the hospital. We ended collection of the data in May 2010. During the study period, 24% of the patients were lost to followup because of transfer to other hospitals or persistent drug-free remission. The median [interquartile range (IQR)] followup period of the patients lost to followup was 30 (IQR 15–68) months. Causes of death were recorded from the patients’ charts and death certificates. Deaths were considered attributable to IIM or not, according to the records.
Prognostic factors
The following prognostic factors were considered: age at the initial presentation, sex, presence of Raynaud’s phenomenon, arthralgia, typical rash, skin ulcers, dysphagia, ILD, myocarditis, and laboratory data of serum CK, ANA and anti-Jo-1 antibodies. We previously reported clinical characteristics of myositis-specific antibodies26, but analysis of those autoantibodies except anti-Jo-1 antibodies was not included in this retrospective study because those autoantibodies were not examined in all patients identified with IIM.
Statistical analysis
Statistical analysis was performed using Prism 4.0c for Macintosh (GraphPad Software Inc., San Diego, CA, USA) and SPSS 17.0 for Macintosh (SPSS Inc., Chicago, IL, USA). For comparisons involving binary data, we used Fisher’s exact tests. Holm adjustment27 was performed for multiple comparisons. For comparisons involving continuous data, we used the Student’s t-test or Mann-Whitney U tests. One-way ANOVA and Kruskal-Wallis tests with corrections by Dunn’s test were applied for multiple comparisons. The survival curves were drawn using the Kaplan-Meier method. The log-rank test was used to determine the statistical significance of the observed differences in survival rates between patient groups. The prognostic factors predicting survival were analyzed using multivariate proportional hazard regression models called Cox regression models.
RESULTS
Clinical characteristics of IIM
Clinical characteristics of the 197 patients are shown in Table 1. Out of those patients, 104 were diagnosed as DM (71 definite, 26 probable, and 7 possible), 72 as PM (41 definite, 21 probable, and 10 possible), and the 21 remaining as clinically ADM. The 176 patients diagnosed with PM/DM were further classified into the 4 subgroups already described. Among the 176 patients, primary DM (42%) was more commonly observed than primary PM (24%). Fourteen patients with SS (37%), 12 with RA (32%), 10 with SSc (26%), and 2 with SLE (5%) were included in OM.
Baseline demographic and clinical characteristics in patients with adult-onset idiopathic inflammatory myopathies.
In comparison with all 5 categories of IIM, dysphagia was most frequently observed in patients with CAM (p < 0.05). Important clinical findings of CAM also included typical skin manifestations such as Gottron’s papules or heliotrope rash (91%). ILD were less frequently found in CAM than in primary DM and OM (p < 0.05). Three of 21 patients with ADM (14%) had malignancy at the diagnosis. These results were consistent with our previous report28. By definition, patients with clinically ADM had low serum CK levels, and almost normal manual muscle testing resulted in differences with each of the other subtypes of IIM (p < 0.05). Serum CK levels were significantly low in primary DM compared to primary PM (p < 0.05). Positive ANA was less frequently detected in primary DM than in OM (p < 0.05). No statistical differences were observed in terms of mean age at diagnosis, sex, and incidence of Raynaud’s phenomenon, arthritis, skin ulcers, myocarditis, and anti-Jo-1 antibodies.
Differences between primary DM and primary PM
We compared clinical characteristics (Table 1) and survival (Figure 1) of primary DM and primary PM, because those findings of other subtypes of IIM such as CAM and OM are potentially determined by the underlying malignancies and complicated collagen vascular diseases, respectively, and not by IIM itself9,10,11.

Survival curves of patients with idiopathic inflammatory myopathies including amyopathic dermatomyositis (A) and patients classified by each subgroup (B). Shown below the figures are number of patients at risk at 30, 60, 90, and 120 months. PM: polymyositis; OM: overlap myositis; DM: dermatomyositis; ADM: amyopathic dermatomyositis; CAM: cancer-associated myositis.
Frequency of ILD (p = 0.0102) and serum CK levels (p = 0.0008) as well as typical rash (p < 0.0001) were different between primary DM and primary PM. Myocarditis was predominantly detected in primary PM, although the result did not reach statistical significance (p = 0.0842). These clinical features observed in primary DM appeared to be applied to clinically ADM in terms of the higher incidence of ILD and lower incidence of myocarditis.
Overall survival and comparison of each subgroup
Global survival rate of the 197 patients with IIM was 85%, 75%, and 67% for 1, 5, and 10 years, respectively (Figure 1). When patients with clinically ADM were excluded, the survival rate was 86%, 77%, and 68% in 1, 5, and 10 years.
Analysis of mortality in each subgroup (Figure 1) revealed that patients with various forms of IIM have different prognoses. The survival rate of patients with CAM was the worst of all the subtypes of myositis (CAM vs primary DM, p = 0.0016), which was consistent with previous studies. Patients with primary DM had significantly lower survival rates compared to patients with primary PM (p = 0.0427). Notably, both patients with clinically ADM and patients with primary DM had accelerated mortality during the first year after initial presentation and a slower mortality during the following years.
Causes of death
Fifty-three patients died during the followup. Out of these, 42 disease-specific deaths (79%) occurred after a median followup of 4.6 (IQR 1.6–25.6) months. The most common cause of disease-specific death was ILD (20 patients), followed by malignancies (15), heart failure (4), and pulmonary hypertension (2). Disease-specific deaths occurred most frequently in CAM (14 patients) followed by ADM (7), primary DM (13), OM (6), and primary PM (2). ILD was the most common cause of death in patients with clinically ADM (5 patients) and primary DM (12) and the death occurred only after the median followup of 1.1 (IQR 0.8–3.4) months. In contrast, death from ILD was rarely seen in primary PM (1 patient) and OM (1). Two patients with OM and 1 patient with primary PM died from heart failure. Two patients with clinically ADM (10%) and 13 with CAM (62%) died from malignancies. The 11 remaining patients died of causes that were not disease-specific. Four patients died from infection and 3 from malignancies that developed later. Other causes of death unassociated with IIM included acute pancreatitis (1 patient) and liver cirrhosis (1 patient).
Prognostic factors and survival
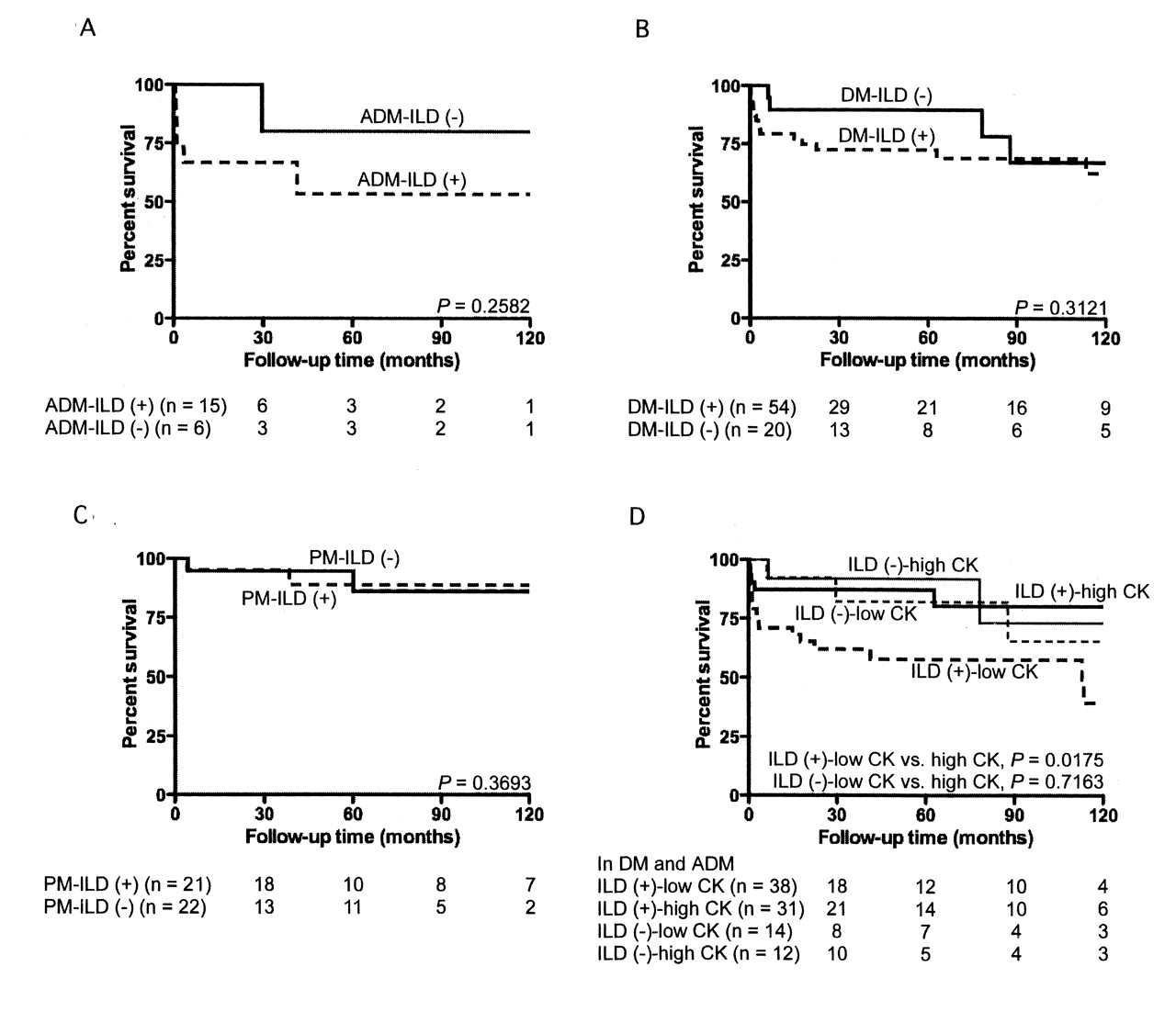
We looked into prognostic factors among clinical manifestations found at initial presentation (Figures 2 and 3). Patients who were age 50 years or older at the time of initial presentation had significantly lower survival in primary DM (p = 0.0022; Figure 2B), but this was not observed in primary PM. In primary DM (p = 0.0209; Figure 2C), OM (p = 0.0049; Figure 2D), and clinically ADM (p = 0.0029), patients who manifested skin ulcers at time of diagnosis had lower survival compared to those patients without them. There was no significant difference between patients with and those without ILD in primary DM, PM, and clinically ADM (Figures 3A, 3B, and 3C). Notably, patients with ILD who had lower serum CK levels (< 500 IU/l) in clinically ADM and primary DM (Figure 3D) had higher mortality, with a 5-year survival rate of 62%, than ILD patients with higher serum CK levels (≥ 500 IU/l; p = 0.0175). Lower serum CK was a poor prognostic factor even when analyzed only among primary DM patients having ILD (p = 0.0206), with a 5-year survival rate of 55%. On the other hand, only 1 patient with primary PM died from ILD. The serum CK level of this patient was > 500 IU/l.

Survival curves in patients with clinically amyopathic dermatomyositis (ADM; A) and primary dermatomyositis (DM; B) sorted by age at initial presentation. Comparison of survival curves in patients with or without skin ulcers in primary DM (C) and overlap myositis (OM; D). Number of patients at risk at 30, 60, 90, and 120 months is shown below the figures.
{kind=link}
{kind=link}
{kind=link}
Survival curves of patients with or without ILD in clinically amyopathic dermatomyositis (ADM; A), primary dermatomyositis (DM; B), and primary polymyositis (PM; C). Comparison of survival curves between patients with lower serum creatine kinase levels (< 500 IU/l) and higher CK levels (≥ 500 IU/l) in patients with ADM and DM, subdivided by the presence or absence of ILD (D). Number of patients at risk at 30, 60, 90, and 120 months shown below the figures. ILD: interstitial lung disease; CK: creatine kinase.
Independent risk factors for death
Cox regression models for patients with IIM are shown in Table 2. In all the patients, older age, skin ulcers, and ILD with low serum CK levels (< 500 IU/l) were independent risk factors for death. When analyzed in primary DM and primary PM separately, older age and ILD with mild serum CK elevation became independent risk factors only in primary DM but not in primary PM.
Cox regression models for patients with idiopathic inflammatory myopathies.
DISCUSSION
We analyzed clinical characteristics, longterm survival, causes of death, and associated risk factors for mortality in patients with IIM. Frequencies of organ involvement, longterm survival, and causes of death were different between patients with primary DM and those with primary PM. The characteristics in clinically ADM appeared to be closer to those in primary DM. Fatal ILD developed more frequently in patients with clinically ADM and primary DM, particularly when serum CK levels were lower. Most of the deaths due to ILD observed in primary DM and clinically ADM occurred within a few months from initial presentation. Consistent with previous studies18,28, each frequency of malignancy and ILD may be similar when compared with DM and ADM. However, it is still unclear, as a recently published population-based study showed, whether the risk of developing malignancies in ADM is as high as that in DM29.
Comparison of survival studies from previous reports is limited30. Although the diagnostic criteria of Bohan and Peter introduced in 197514,15 had been used in most studies (except for those by Medsger, et al2 and Hochberg, et al31, which had started before the criteria were established), these longterm series had different inclusion criteria, making direct comparison difficult9,30. Moreover, survival in IIM varies widely depending on the inclusion or exclusion of CAM (Table 3)11. Survival studies according to each subgroup with sufficient numbers of patients were not available until the report from Hungary by Danko, et al in 20049. More recently, a Spanish group reported the longterm survival of IIM according to each subgroup of IIM11. Even if a survival study of IIM was performed based on Bohan and Peter’s criteria and classification14,15, problems still existed. One problem regarding this classification that affected the IIM survival rate is that OM was too loosely defined. Dalakas and Hohlfeld insisted that only DM, and not PM, truly overlaps only with SSc and mixed connective tissue disease32. If this definition were applied to patients with OM in our study, 34 out of 38 patients (89%) with OM would be classified as primary PM.
With those problems in mind, we compared longterm survival of IIM with previous reports by analyzing patients with IIM according to subgroups as well as a whole group of patients (Table 3).
The survival rate was lower in patients with DM than in patients with primary PM in our study. This was in contrast to reports from Europe9,10,11, which showed no difference in survival rates between them. Lower survival rates of patients with ADM and primary DM was mainly due to severe ILD, and this may be a specific feature in eastern Asia. ILD was the main cause of death in clinically ADM as well as in primary DM. More patients died from ILD than from malignancies. Progressive ILD in ADM has been reported mainly in such Asian countries as Japan, South Korea, China, and Taiwan13,24,33,34,35, while Cottin, et al reported a benign course of ILD in European patients with ADM36. In fact, previous studies from eastern Asia supported our observation that the survival rate was lower in patients with ADM-associated and DM-associated ILD than in those with PM-associated ILD37,38 and steroid-resistant ILD developed mostly in patients with PM or DM with accompanying low CK levels39. Only 1 patient with primary PM and 2 with OM died from heart failure in our study, which contrasted with the finding that cardiac involvement was the most common cause of death except malignancies and was one of the factors associated with mortality among the European population with IIM9,11.
Previous case reports suggested that skin ulcers, one of the manifestations of cutaneous vasculitis, might be poor prognostic signs of DM and were associated with pneumomediastinum40 or fatal ILD41. Our study showed that patients with clinically ADM and primary DM who had skin ulcers at the initial presentation had significantly lower survival rates than those without them. Indeed, 33% of primary DM and 100% of clinically ADM with skin ulcers died from fatal ILD. On the other hand, skin ulcers in patients with OM could be more associated with severity of other collagen vascular diseases such as SSc or SS.
One limitation of our study is that it is a retrospective analysis conducted in a single institute. In addition, 24% of the patients were lost to followup, so the data on survival may be somewhat biased. Serological screenings were not performed, except for ANA and anti-Jo-1 antibodies.
ILD was the most common cause of death in IIM in our patient population, especially in patients with primary DM who had lower serum CK levels and clinically ADM. Considering that death from ILD in patients with primary DM and clinically ADM occurred only after a median of 1.1 months from the initial visits, evaluation of ILD should be made at the disease onset. The future direction of study should include establishing a therapeutic strategy for ILD to improve survival of patients with IIM.
The treatment approach for ILD has changed over time. Aggressive use of immunosuppressive agents such as cyclophosphamide42,43, cyclosporine39, or tacrolimus44 for the treatment of steroid-resistant ILD has been introduced and appears to have favorable outcomes. The effect of those aggressive treatments on survival needs to be evaluated.
Acknowledgment
We appreciate advice from Dr. Takahiko Ueno and Dr. Shinobu Tatsunami on statistical analysis, and from the many patients and their referring physicians at the Division of Rheumatology and Allergology, St. Marianna University Hospital. This study would not have been possible without their cooperation. We are deeply grateful to Dr. Minoru Satoh (University of Florida, Gainesville, FL, USA) for valuable contributions to our studies.
- Accepted for publication March 17, 2011.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.