

Abstract
The risk of developing active tuberculosis (TB) is higher in patients taking immunosuppressive drugs, either as a result of reactivation of a latent TB infection (LTBI) or following a new infection with Mycobacterium tuberculosis (Mtb). We discuss the pathogenesis and spectrum of Mtb infection in light of its implication for the management of patients following biologic regimens. Among recent findings, during LTBI, Mtb can persist in the host for decades, localizing in many tissues and assuming different metabolic states that protect the bacilli from the harsh host immune defenses. Despite the strong host T cell response against Mtb, the bacilli may also replicate and multiply in vivo, and any event impairing immune function may lead to active and uncontrolled bacteria replication and active disease. The classic dichotomy between active and latent disease is being reconsidered in favor of a continuous and dynamic spectrum extending from infection to disease that can coexist in the same individual. This TB spectrum results from the dynamic interaction between the host immune system and the bacilli and can be maintained in equilibrium for decades, although treatments affecting the host immune cells may result in disease reactivation.
Patients taking immunosuppressive therapy such as corticosteroids and anti-tumor necrosis factor (TNF) biologics show increased risk of developing active tuberculosis (TB), either as a result of reactivation of a latent infection or progression of primary infection. Management of Mycobacterium tuberculosis (Mtb) infection in these patients is crucial to prevent active, often complicated, disease. In this context, a summary of the most recent advances in the biology of Mtb infection, with a focus on latent TB infection (LTBI), will be instrumental to identifying and designing effective management of these patients.
Early Events Following Mtb Infection
Once Mtb is inhaled by a human host it enters the alveolar space, where it is engulfed by professional macrophages that can effectively kill the bacilli, aborting the infectious process. It is estimated that in 20–50% of subjects exposed to Mtb, the bacilli can resist these innate immune defenses and begin to replicate within alveolar macrophages, local dendritic cells, interstitial macrophages, and possibly epithelial cells1. Compared to other pathogens, Mtb has a unique ability to delay initiation of adaptive immune responses, both in humans and in mice, probably because Mtb-infected dendritic cells fail to migrate from the infected lung focus to the local draining lymph nodes, in which T cell priming takes place, for a period of at least 2 weeks2,3,4. This delay may allow Mtb to establish a critical mass of bacilli before adaptive immunity kicks in and controls infection5,6. The emergence of the adaptive immune response results in a cellular infiltrate that organizes in a granuloma, which is the hallmark of Mtb infection. In 90–95% of cases, this cellular immune response can effectively control bacteria replication, preventing further tissue damage and the emergence of overt signs or symptoms of disease, a condition termed LTBI. When this adaptive primary immune response fails to properly control bacterial replication and limit tissue damage, active disease ensues with the classic clinical signs of TB disease (primary progressive TB). Subjects with LTBI remain asymptomatic and usually never develop active TB, although the likelihood of developing disease is ∼5–10% during a lifetime (reactivation TB), with half of this risk concentrated in the first 2 years after infection. The immunological and molecular mechanisms underlying TB reactivation are not clear, although it is well known that several conditions inducing immunosuppression may increase the likelihood of TB reactivation.
Where Does Mtb Reside During Latency?
For a long time it has been proposed that Mtb persists in old tuberculous lesions such as healed pulmonary and tracheo-bronchial granulomas, where bacilli were thought to remain encapsulated in these eventually calcified lesions, protected by the host cellular immune response7. In this scenario, following Mtb entry and bacilli replication, the host immune response prevents bacterial dissemination and spread from the site of infection, containing the bacilli in this primary lesion, termed the Ghon complex8. The Ghon complex was considered the “sanctuary” of Mtb, and destruction of this old lesion would result in bacteria replication and spread, tissue damage, and TB reactivation.
Several studies attempted to investigate this issue by analyzing pulmonary lesions at necropsy of patients who had died of causes other than TB. Encapsulated lesions isolated from LTBI subjects were shown to be microbiologically sterile (Mtb could not be cultured) and a higher bacterial viability was observed in fibrotic and caseous lesions or from tissue homogenates from unaffected portions of the lung7. More recently, using sensitive molecular techniques on tissue isolated from necropsy, it was demonstrated that mycobacterial DNA could be found in lung tissue from LTBI subjects without histological evidence of TB lesions, and DNA could be detected intracellularly in macrophages and other cell types such as type II pneumocytes, endothelial cells, and fibroblasts9. Using a similar approach, Neyrolles, et al10 were able to demonstrate that Mtb could be found in fat tissue surrounding the kidneys, stomach, lymph nodes, heart, and skin, where it is found intracellularly in adipocytes. Hence, fat tissue and adipocytes might constitute a vast reservoir where Mtb could persist for long periods of time, avoiding recognition by the host immune response and killing by antimicrobials10.
This evidence indicates that during LTBI, tubercle bacilli reside in many different tissues that are not associated with the site of primary infection. It follows that immediately after entry, once the bacilli start multiplying in alveolar macrophages and infecting nearby cells, Mtb spreads to other tissues11. Some infected macrophages migrate through the lymphatics to enter the bloodstream and to seed secondary lesions12. Replication of Mtb in macrophages at the site of primary infection causes macrophage death by necrosis13, which spreads live bacilli that can infect epithelial cells and disseminate hematogenously to potentially reach any organ14. Indeed, Mtb has been found to infect type II pneumocytes in lung alveoli in the early steps of infection15, and a mycobacterial surface protein (HBHA) was shown to mediate adhesion and invasion of epithelial cells, thereby promoting bacilli dissemination16,17. Dissemination from the site of primary infection is therefore a crucial step in TB pathogenesis and allows the bacteria to reach potentially any organ where they can persist for a long time and eventually reactivate and cause disease, particularly in more susceptible tissues sites such as the upper lung lobes14.
Metabolic States During Infection
Investigation of latent infection in humans has proven difficult, and the classical animal models used to study TB pathogenesis, such as the mouse, guinea pig, and rabbit, do not mimic latent infection18, and only recent studies in nonhuman primates have shed light on this elusive step of Mtb infection19. One important issue to address is to determine the Mtb metabolic status during LTBI. It was shown that bacteria inside some old lesions may be found to be viable but do not divide, which is referred to as a dormant state. Conditions that are found in macrophages or in granulomas, such as low oxygen tension, nutrient starvation, and the presence of nitric oxide and carbon monoxide, induce dormancy20. Bacteria may be found in a viable but nonculturable state, which may limit the possibility to isolate Mtb from old lesions or other biological samples. During the dormant state bacteria are capable of switching toward an anaerobic metabolism, through the downregulation of central metabolism, activation of the glyoxylate cycle, and other alternative metabolic pathways, including triacylglycerol biosynthesis, which is known to play an important physiological role during the dormant state18,21. Induction of stress proteins and ultrastructural modifications on the mycobacterial cell wall characterize the dormant state, and these complex changes are orchestrated by the dormancy survival regulon, which controls the Mtb hypoxic response through the regulation of more than 50 transcripts22. The toxin-antitoxin loci, which are present in a very high number in the Mtb genome compared to other bacteria, encode proteins thought to bind RNA and exert a bacteriostatic effect during hypoxia and starvation, which facilitate dormancy23.
Dormant bacteria may reverse their stable nonreplicating state into a metabolically active growing population in a process termed resuscitation, which involves specific proteins termed resuscitation promoting factors (Rpf)18,24. Mtb encodes 5 Rpf proteins, which are capable of promoting bacterial growth, as demonstrated also in ex vivo studies, where Rpf increased the recovery of Mtb from the sputum of patients with active TB25,26. Rpf are thought to induce bacterial growth by digesting or modifying the peptidoglycan and triggering a signaling cascade that activates metabolism18.
The differential pattern of susceptibility of dormant and active bacilli against the classical antituberculous drugs illustrates the physiological differences between these 2 metabolic states. Dormant bacilli are phenotypically resistant to isoniazid, which is known to be active only against growing bacilli27, probably because these nonreplicating cells can resist inhibition of mycolic acid biosynthesis caused by isoniazid. Rifampin, an RNA polymerase inhibitor, is active against actively replicating bacilli but is known to be effective against dormant bacteria as well, suggesting that these cells are not inert but instead maintain a certain degree of transcriptional activity28. Metronidazole, a drug active against anaerobic bacteria, is bactericidal against Mtb under hypoxic conditions in a nonreplicating dormant state29 but has no apparent activity for growing bacilli in aerobic conditions30. Pyrazinamide (PZA), a very effective drug against Mtb and one that plays an important role in shortening the duration of TB chemotherapy, has no activity against growing bacilli under normal culture conditions31. It has been demonstrated that pyrazinoic acid, the product of the intracellular hydrolysis of PZA by pyrazinamidase, binds the ribosomal protein S1 inhibiting trans-translation, which is an essential process that frees scarce ribosomes in nonreplicating organisms, thereby explaining the peculiar ability of PZA to eradicate persisting, dormant bacilli32. These results underscore the effect that the different Mtb metabolic states have on drug susceptibility, pointing out that an effective drug regimen must target these different populations of bacilli simultaneously.
The Spectrum of TB
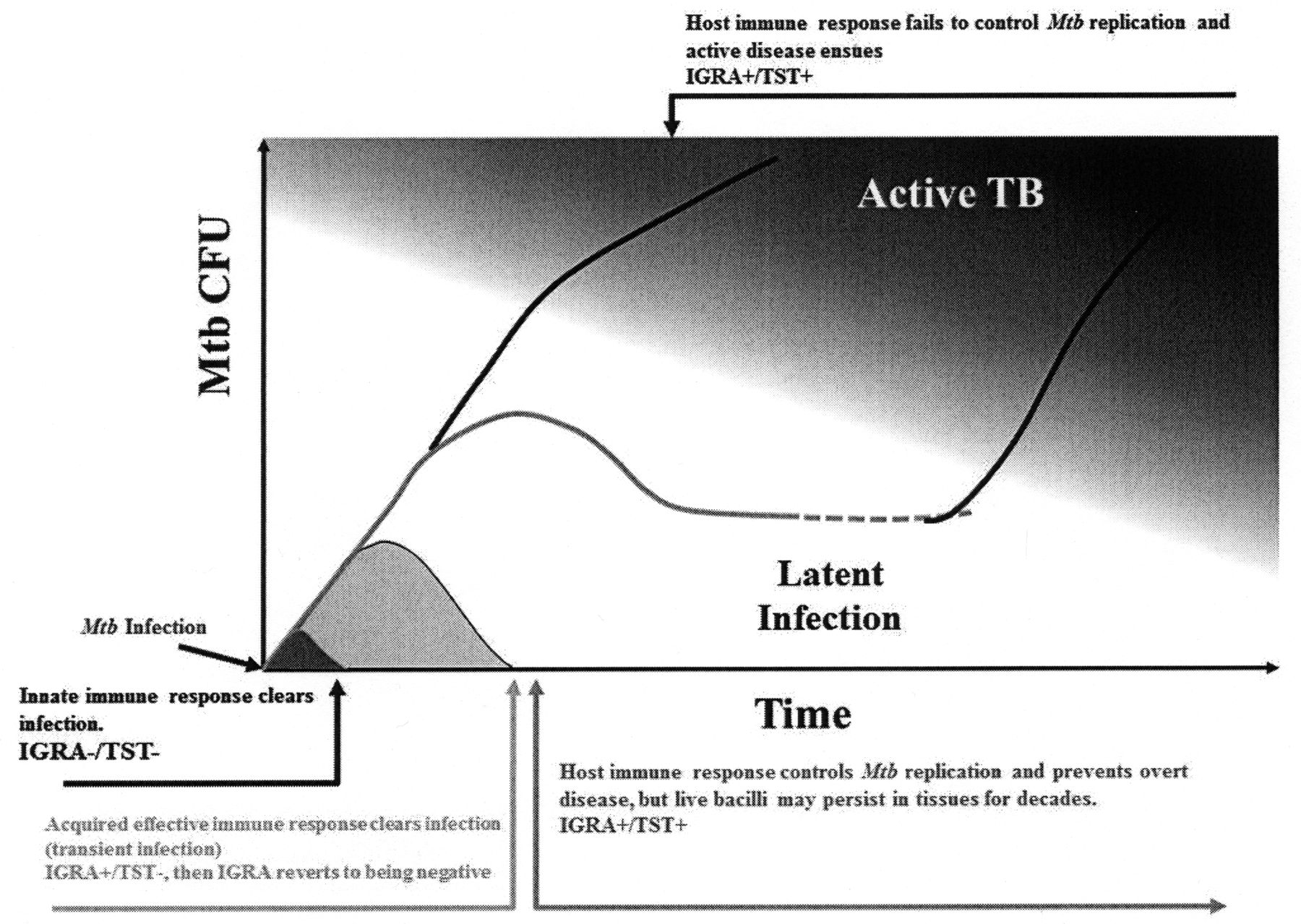
Dormant and actively replicating bacilli are concomitantly present during Mtb infection, with the balance among these 2 populations skewed toward one or the other, depending on the clinical status33. This new view of Mtb infection comes from several observations obtained in active TB patients, LTBI subjects, and experimental studies on nonhuman primate models of infection19. During active TB, it is possible to detect sterile tissue, solid, necrotic, or caseous hypoxic lesions containing variable numbers of bacteria, or liquefied cavities with massive loads of replicating bacilli33. These diverse lesions coexist simultaneously in the same patients with TB, and positron emission tomography and computer tomography imaging illustrated that these lesions respond differently to chemotherapy, indicating that they represent distinct bacterial subpopulations in different microenvironments33. The heterogeneity of TB lesions has also been observed in LTBI subjects33,34, suggesting that also latent infection can be depicted as a broad spectrum of conditions that overlap with those seen in active disease. Some subjects show only the remnant of a waning infection, while others show a slowly progressing form of the disease, or a chronic nonprogressing infection33. This spectrum of conditions typical of latent infection has also been confirmed in the TB monkey model, with some animals, termed “percolators,” showing subclinical TB35. Interestingly, the estimated Mtb mutation rate in monkeys was similar during active TB and latent infection, suggesting that during latent infection Mtb is not simply in a dormant persistent, nonreplicating condition, but that it actively replicates36,37. These new lines of experimental evidence provide a new dynamic model of latency. Following primary infection and control of bacterial replication by the adaptive immune response, bacteria can reside and persist in different tissues in a dormant state, warranting enhanced resistance from the antimicrobial activity of the host immune response. Some of these dormant bacteria resuscitate and start actively replicating, functioning as scouts because they test the environment for acceptable growth conditions18. In an immunocompetent host, these replicating bacilli are likely killed, and the dormant population predominates. When the host immune response fails to properly control this metabolically active population, bacterial replication is not controlled and reactivation and secondary active TB may develop. Isoniazid is effective in treating LTBI because it targets these Mtb scouts, depleting the population of metabolically active bacilli and preventing progression to active TB disease38.
Hence, the classic dichotomy between active disease and LTBI has been reconsidered in favor of a continuous and dynamic spectrum of conditions extending from infection to disease19,33,39 (Figure 1). The host immune response can prevent Mtb infection by killing the bacilli immediately after entry as a result of the innate immunity, or eventually through an effective adaptive immune response, which can clear the infection (transient infection)40. The immunological events associated with bacilli clearance are not known, although it has been proposed that the ability to clear infection might be associated with a low dose of infection41. The possibility that T cell adaptive immunity may play a role in this clearance has been suggested by studies in contacts, where subjects with a positive interferon-γ release assay (IGRA) result later reverted to negative and never converted to tuberculin skin test positivity40. When Mtb survives these host immune responses, then latent infection takes place as highlighted by the typical conversion to purified protein derivative and positivity to the IGRA test. That the spectrum of LTBI in humans may resemble what is observed in monkeys35 and the observation that during LTBI in humans the T cell response measured by IGRA may dynamically change over time support this hypothesis42. It is plausible that the persisting T cell response controls bacterial replication and prevents disease progression or reactivation, although the mechanisms are only partially known. It remains still unresolved, for instance, why many patients develop active TB disease despite having a normal T cell function43 as classically highlighted by the high incidence of active TB in the young adult population, which is still observed in TB-endemic countries44. However, events that impair the host T cell response, such as certain infections (human immunodeficiency virus), malnutrition, or treatment with immunosuppressive drugs may result in uncontrolled replication of bacilli and progression of TB, which in some cases may present as a disseminated and fatal disease.

The natural history of tuberculosis (TB) infection. CFU: colony forming units; IGRA: interferon-γ release assay; TST: tuberculin skin test; Mtb: Mycobacterium tuberculosis.
Immunological Mechanisms Underlying Increased Risk of TB Infection and Reactivation in Patients Undergoing Biological Therapies
In the context of the continuous spectrum of TB infection, the maintenance of the correct immunological homeostasis during latent infection is of great importance. Any drug treatment affecting the host cell response has an effect on the ability of the host to keep bacteria replication under control in a dynamic process, as classically highlighted by the treatment with corticosteroids or anti-TNF biologics45.
TNF is required for restricting mycobacterial growth in vivo, and studies carried out in animal models are helping to unravel the immunological mechanisms involved46. Lack of TNF in mice infected with Mtb was associated with impaired protective immune response, with granulomas lacking proper cellular organization and characteristic architecture47. Blockade of TNF signaling may have multiple effects on the maintenance of granulomas, such as increased non-apoptotic death of macrophages spurring mycobacterial replication; moreover, imbalance of chemokine and chemokine receptor expression in TNF-depleted, Mtb-infected host influences adhesion-protein expression, which in turn may decrease recruitment or increase egress of lymphocytes, macrophages, or dendritic cells48. Recently, in mouse and human granulomas, specific populations of macrophage-derived epitheliod cells expressing the T cell receptor-αβ (TCR-αβ) were found at the interface of host/pathogen interaction. TNF blockade suppresses macrophage-TCR-αβ expression, resulting in granuloma disorganization and mycobacterial spread49.
CD4 and CD8 T lymphocytes are known to play an important role in controlling Mtb replication, and increased T cell apoptotic death in TNF-depleted hosts is followed by increased bacterial replication due to altered bacterial gene expression and mycobacterial spread48. Interestingly, anti-TNF immunotherapy with infliximab was shown to specifically deplete a specific population of CD8 T lymphocytes expressing granulysin, which are capable of deploying a strong antimicrobial response. These CD8 T cells (TEMRA cells) express TNF on their surface, and binding of anti-TNF triggers a complement mediated death that leads to mycobacterial growth and spread50,51. The uncovering of this mechanism underscores the role of CD8 T cells in controlling Mtb replication, particularly during LTBI52, and provides a model to explain the increased risk of TB reactivation in patients treated with anti-TNF (infliximab) compared to those treated with the soluble TNF receptor (etanercept)45.
Footnotes
-
Supported by the Italian MIUR (PRIN grant number 2008 Y8RZTFMS).








{kind=link}