

Abstract
Objective. Evaluate safety and efficacy of intravenous (IV) golimumab (GOL) in patients with active ankylosing spondylitis (AS) through 1 year.
Methods. A total of 208 patients were randomized to IV infusions of GOL 2 mg/kg (n = 105) at weeks 0, 4, and every 8 weeks thereafter or placebo (n = 103) at weeks 0, 4, and 12, then crossover to GOL at weeks 16, 20, and every 8 weeks thereafter through Week 52. Efficacy was assessed using the Assessment of Spondyloarthritis international Society (ASAS) criteria, the Ankylosing Spondylitis Disease Activity Score (ASDAS), the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and the Bath Ankylosing Spondylitis Functional Index (BASFI). Health-related quality of life was assessed using the AS Quality of Life (ASQoL) index. Efficacy and safety were monitored through Week 52 and Week 60, respectively.
Results. The primary endpoint (ASAS20) and all controlled endpoints at Week 16 were achieved. At Week 52, 69.5% and 65.0% of patients in the GOL group and placebo crossover group, respectively, achieved an ASAS20; 56.2% and 51.5% achieved an ASAS40; 56.2% and 55.3% achieved a BASDAI50; 24.8% and 24.3% achieved ASAS partial remission; and 25.7% and 26.2% met ASDAS inactive disease criteria (all last observation carried forward). Mean changes from baseline to Week 52 in BASFI and ASQoL scores were similar between the GOL group and the placebo crossover group (BASFI: −2.7 and −2.6; ASQoL: −5.5 and −5.4). Through Week 60, 55.4% of all GOL-treated patients had ≥ 1 adverse events (AE); 3.4% had ≥ 1 serious AE.
Conclusion. Efficacy was maintained through 1 year with IV GOL 2 mg/kg among patients with active AS. AE were consistent with the known safety profile of GOL.
Ankylosing spondylitis (AS) is an immune-mediated disease characterized by inflammatory back pain and progressive spinal stiffness1. As the disease progresses, there is a decrease in spinal mobility and physical function, and an increase in disability. This can lead to substantial impairment in health-related quality of life (HRQOL)2. Current treatment recommendations for AS include the use of nonsteroidal antiinflammatory drugs (NSAID), with initiation of anti–tumor necrosis factor (TNF) agents for patients with active disease despite treatment with NSAID3.
Shared decision making with patients can improve treatment satisfaction4 and is part of the treatment discussion between patients and healthcare providers. Therapies are available with a variety of dosing frequencies and routes of administration, and patients may have preferences related to these factors. In a study of patients with rheumatoid arthritis who were receiving anti-TNF therapy, patients who preferred subcutaneous (SC) therapy indicated a preference for self-administration at home, while patients who preferred intravenous (IV) therapy expressed a preference for receiving therapy in the presence of a hospital or physician5. Frequency of administration was cited as a preference for patients receiving IV therapy, but not SC therapy5.
The GO-ALIVE trial evaluated the safety and efficacy of IV golimumab (GOL), a monoclonal anti-TNF antibody, in patients with AS6. Through the placebo-controlled period of the GO-ALIVE trial (Week 16), patients who received IV GOL 2 mg/kg had greater improvements in the signs and symptoms of AS than did patients receiving placebo, and these improvements were maintained through Week 286. In addition, adverse events (AE) that occurred through Week 28 were consistent with the known safety profile of IV GOL and other anti-TNF agents6. Clinical efficacy, safety, and pharmacokinetic results through 1 year of the GO-ALIVE trial are reported herein.
MATERIALS AND METHODS
Patients and study design.
Detailed eligibility criteria and study design were previously reported6. Adults with a diagnosis of AS (“definite” using the modified New York criteria) for ≥ 3 months with signs of active disease and inadequate response or intolerance to NSAID were eligible for inclusion in GO-ALIVE. Up to 10% of the study population could have complete ankylosis of the spine. Eligible patients were randomly assigned to receive IV infusions of placebo at weeks 0, 4, and 12 or GOL 2 mg/kg at weeks 0, 4, and every 8 weeks thereafter through Week 52. Patients in the placebo group crossed over to receive GOL 2 mg/kg at weeks 16, 20, and every 8 weeks thereafter through Week 52.
Stable doses of methotrexate (MTX; ≤ 25 mg/week), sulfasalazine (SSZ), hydroxychloroquine (HCQ), NSAID, other analgesics, and low-dose oral corticosteroids (dose equivalent to ≤ 10 mg prednisone/day) were permitted for patients who were receiving these medications at baseline. Patients could not have received systemic disease-modifying antirheumatic drugs other than MTX, SSZ, or HCQ within 4 weeks of the first study agent administration. Up to 20% of the study population could have received a prior anti-TNF therapy other than GOL; these patients could not have discontinued the prior anti-TNF agent because of primary treatment failure and could not have received anti-TNF therapy within 3 months of the first study agent administration (except for etanercept within 6 weeks).
Assessments
As previously reported, the primary endpoint was the proportion of patients with an improvement of ≥ 20% in the Assessment of Spondyloarthritis international Society criteria (ASAS20 response) at Week 166. The proportions of patients achieving an ASAS20 response, ASAS40 response, ASAS partial remission (score < 2), and ASAS 5/6 response7 were evaluated through Week 52. In addition, disease activity was also assessed using the Ankylosing Spondylitis Disease Activity Score (ASDAS; inactive disease, score < 1.3; major improvement, decrease ≥ 2.0; and clinically important improvement, decrease ≥ 1.1)8,9 and the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)10. Maintenance of response at Week 52 was determined among those patients who had an ASAS20 response, ASAS40 response, ASDAS inactive disease, or BASDAI50 response at Week 16. Physical function was assessed using the Bath Ankylosing Spondylitis Functional Index (BASFI)11. Enthesitis was assessed using the University of California San Francisco enthesitis index12.
HRQOL was evaluated using the 36-item Medical Outcomes Study Short Form-36 (SF-36) physical component summary (PCS) and mental component summary (MCS) scores13 and the AS Quality of Life (ASQoL) score14. Improvements in spinal mobility were assessed using the Bath Ankylosing Spondylitis Metrology Index (BASMI)15.
AE were monitored through Week 60. Serum samples were collected to measure GOL concentrations and evaluate for the presence of antibodies to GOL using a highly sensitive, drug-tolerant, enzyme immunoassay (EIA).
Statistical methods
Clinical efficacy results through Week 52 were summarized by randomized treatment group (intent-to-treat) using descriptive statistics (counts and percentages for discrete variables; means and SD for continuous variables). No formal comparisons were performed for timepoints after Week 16, when patients in the placebo group crossed over to GOL. Missing data were imputed using last observation carried forward methods. For dichotomous composite endpoints with all components missing, nonresponder imputation was used through Week 52. No treatment failure rules6 were applied after Week 16.
AE through Week 60 were summarized by the treatment actually received and included all patients who received at least 1 administration of GOL (all GOL group). Pharmacokinetic and immunogenicity analyses included all patients who had received ≥ 1 GOL administration and had ≥ 1 post-GOL administration serum sample to assess serum GOL concentration.
RESULTS
Patients
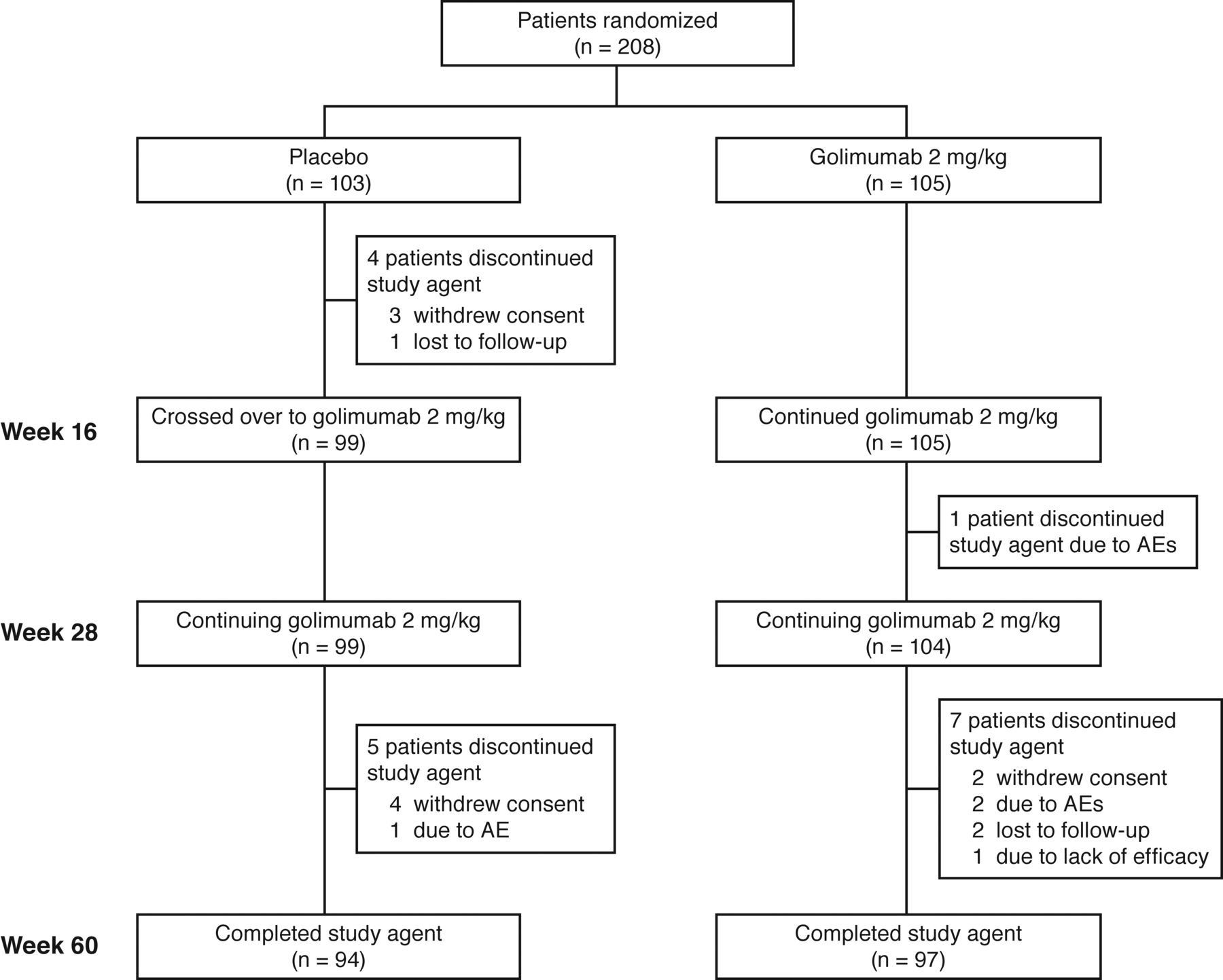
Baseline characteristics and patient disposition through Week 28 have been previously reported6. Briefly, 208 patients were randomized to receive placebo (n = 103) or GOL 2 mg/kg (n = 105). Baseline demographic and disease characteristics were well balanced between the treatment groups6. Through Week 60, nine patients in the placebo group (4 prior to crossover to GOL at Week 16 and 5 after Week 16) and 8 patients in the GOL group discontinued study treatment (Figure 1). Overall, the most common reasons for discontinuing the study agent throughout the study were withdrawal of consent (n = 9) and AE (n = 4).

Patient disposition through Week 60. AE: adverse event.
Clinical efficacy
The primary endpoint was achieved, with 73.3% of patients in the GOL group achieving an ASAS20 response at Week 16 compared with 26.2% in the placebo group6. As previously reported6, greater proportions of patients in the GOL group achieved an ASAS40 response, ASAS partial remission, ASAS 5/6 response, BASDAI50 response, BASDAI70 response, and ASDAS inactive disease compared with the placebo group at Week 16. The proportion of patients who had a clinically important improvement in ASDAS from baseline was greater in the GOL group at Week 16 (82.7% vs 22.5%; p < 0.001); response rates for patients with a clinically important improvement in ASDAS were similar in the 2 treatment groups at weeks 28 (GOL: 76.9%; placebo crossover: 71.6%) and 52 (GOL: 76.0%; placebo crossover: 71.6%; Table 1). Following placebo crossover to GOL at Week 16, the proportions of patients achieving an ASAS20, ASAS40, ASDAS inactive disease, or BASDAI50 response in the placebo group approached those observed in the GOL group, as early as Week 20 (4 weeks after crossover to GOL)6 and remained steady through Week 52 (Figure 2) in both treatment groups. Additionally, among those patients who achieved an ASAS20, ASAS40, ASDAS inactive disease, or BASDAI50 response at Week 16, most maintained this response at Week 52 (Table 2). In addition, mean changes from baseline in BASFI and BASMI scores were similar between the treatment groups at Week 52, when all patients had been receiving GOL after the placebo crossover at Week 16 (Table 1).

Proportions of patients achieving an (A) ASAS20 response, (B) ASAS40 response, (C) BASDAI50 response, and (D) ASDAS inactive disease through Week 52. Patients randomized to placebo crossed over to golimumab 2 mg/kg at Week 16 (dotted line). Missing data were imputed using last observation carried forward. For dichotomous composite endpoints with all components missing, nonresponder imputation was used through Week 52. ASAS20/40: ≥ 20%/40% improvement in Assessment of SpondyloArthritis international Society criteria; ASDAS: Ankylosing Spondylitis Disease Activity Score; BASDAI50: 50% improvement in Bath Ankylosing Spondylitis Disease Activity Index.
Efficacy at Week 52a.
Maintenance of efficacy from Week 16 to Week 52a.
Among patients with enthesitis at baseline, the mean change from baseline in enthesitis score was also maintained from Week 28 through Week 52 in both treatment groups (GOL: −3.8; placebo crossover: −3.6; Table 1). A greater proportion of patients in the GOL group had an enthesitis score of 0 (resolution of enthesitis) at Week 16 compared with placebo (43.7%. vs 14.1%; p < 0.0001). After placebo crossover to GOL, 47.1% in the GOL group and 42.4% of patients in the placebo crossover group had resolution of enthesitis at Week 28, and 59.8% and 42.4%, respectively, had resolution of enthesitis at Week 52.
Improvements in SF-36 PCS/MCS and ASQoL scores were greater for patients in the GOL group compared with placebo at Week 166. These improvements were maintained at Week 52 and were similar between the 2 treatment groups (Table 1).
AE
Through Week 60, 204 patients received at least 1 administration of GOL, including 99 patients who crossed over from placebo at Week 16 and 105 patients who were randomized to GOL at baseline. AE that occurred through Week 16 (placebo-controlled period) and through Week 28 have been previously described in detail6. Cumulative safety results through Week 60 are presented in Table 3. Among all GOL-treated patients, 55.4% had ≥ 1 AE through Week 60. The most common type of AE was infection (n = 67, 32.8%). The most common AE were nasopharyngitis (11.8%), upper respiratory tract infection (7.4%), and alanine aminotransferase (ALT) increase (5.9%). Three (1.5%) GOL-treated patients had a serious infection; one (pneumonia) occurred in a patient in the GOL group before Week 16 and was previously reported6. The other 2 serious infections occurred after Week 28: one patient had appendicitis, and one patient had pulmonary tuberculosis (Ukraine; screened negative for tuberculosis by QuantiFERON-TB Gold test and chest radiograph). Four patients discontinued the study agent owing to an AE; all had been treated with GOL. One patient [increased ALT and increased aspartate aminotransferase (AST)] was previously reported6, and 3 discontinued after Week 28 (increased ALT, pulmonary tuberculosis, and rash).
Adverse events (AE) through Week 60.
A total of 8 serious AE (SAE) occurred; 2 occurred through Week 16 (pancreatitis and pneumonia, both in the GOL group)6. After Week 16, six SAE occurred: appendicitis, pulmonary tuberculosis, sinus tachycardia, nonalcoholic steatohepatitis, wrist fracture, and Henoch-Schönlein purpura (same patient who had pneumonia). There were no opportunistic infections, demyelinating events, malignancies, or deaths during the study. In addition, there was a nonserious report of acute hepatitis B at Week 60 in a patient who screened negative for hepatitis B at baseline.
Cumulatively through Week 60, sixty-five of the 204 GOL-treated patients who had an ALT level in the normal range at baseline had a maximum postbaseline value above the upper limit of normal (ULN). Of these patients, 63 had an ALT level < 3 × ULN, and 1 patient had an ALT level ≥ 3 to < 5 × ULN. One patient had an ALT level ≥ 8 × ULN (the patient diagnosed with acute hepatitis B infection at Week 60).
Additionally, 35 of the 204 GOL-treated patients had an AST level at baseline that was within the normal range and a maximum postbaseline level that was above the ULN. All increases were < 2 × ULN, except for the patient with acute hepatitis B who had a postbaseline AST level ≥ 5 to < 8 × ULN.
Among the subset of 21 GOL-treated patients who received tuberculosis prophylaxis, 10 had an elevated postbaseline ALT level. Nine of these patients had an ALT level < 3 × ULN; 1 patient had an ALT level ≥ 8 × ULN (patient with acute hepatitis B).
A total of 1500 GOL infusions were administered. Four infusion reactions, defined as an AE that occurred during the infusion or within 1 h following the end of study agent administration, were reported in 3 patients, all during the placebo-controlled period6. None was considered serious or severe, and none led to discontinuation of study agent. No additional infusion reactions occurred through Week 60.
Pharmacokinetics and immunogenicity
Median trough serum GOL concentrations reached steady state by Week 12 in the GOL group6 and were maintained through Week 52; patients in the placebo group who crossed over to GOL at Week 16 had similar median serum GOL concentrations from Week 20 through Week 52. Through Week 52, 203 patients received ≥ 1 GOL infusion and had ≥ 1 postbaseline serum sample available for evaluating immunogenicity using the drug-tolerant EIA method. Of these, 41 (20.2%) tested positive for antibodies to GOL, 12 of whom tested positive for neutralizing antibodies. Given the higher sensitivity of the drug-tolerant EIA method, most patients who tested positive had a low titer (< 1:1000). Patients with titers < 1:1000 typically had detectable trough GOL concentrations, while the 2 patients with titers ≥ 1:1000 had trough concentrations below the lower limit of quantification at weeks 20, 36, and 52. No infusion reactions occurred in patients who tested positive for antibodies to GOL. Among GOL-treated patients who tested negative for antibodies to GOL (n = 157), 73.2% achieved an ASAS20 response and 59.2% achieved an ASAS40 response at Week 52; among those who tested positive for antibodies to GOL (n = 39), 64.1% achieved an ASAS20 response, and 48.7% achieved an ASAS40 response. There was no apparent relationship by antibody titers; however, the relatively small number of patients in each titer group limits interpretation (data not shown).
DISCUSSION
In the phase III GO-ALIVE trial of patients with active AS, the primary endpoint was achieved at Week 16 with 73.3% of patients randomized to receive GOL achieving an ASAS20 response compared with 26.2% of patients randomized to placebo6. Following placebo crossover to GOL at Week 16, patients in the placebo crossover group achieved efficacy responses similar to those observed in the patients who had received GOL from baseline, as assessed by ASAS criteria and BASDAI and ASDAS scores. Among patients in the placebo crossover group, response to GOL at Week 20, four weeks after patients in this group initiated GOL therapy, was generally similar to that observed among patients who had been receiving GOL from baseline. This rapid improvement may be at least partially attributed to the placebo response occurring through Week 16 in these patient-driven outcomes, with further improvement upon switching to GOL. A consistent, high level of response was observed in both treatment groups through 1 year. GOL concentrations were also maintained at steady state in both treatment groups through 1 year. The proportions of patients who achieved ASAS20 and ASAS40 responses at Week 52 were slightly lower for patients who were positive for antibodies to GOL compared with those who tested negative; however, a positive antibody-to-GOL status did not preclude clinical response. The drug-tolerant EIA method is highly sensitive; most of the patients who tested positive for antibodies to GOL had low titers and detectable trough GOL concentrations. Two patients had titers ≥ 1:1000, and these patients had undetectable trough GOL concentrations beyond Week 20.
Improvements in HRQOL were also maintained through 1 year with IV GOL. At Week 52, 69.5% of all GOL-treated patients achieved an ASAS20 response and 56.2% achieved an ASAS40 response. Of note, most patients who had an ASAS20, ASAS40, ASDAS inactive disease, or BASDAI50 response at Week 16 were also in response at Week 52.
Few patients discontinued study treatment through 1 year, with over 90% of patients completing study infusions through Week 52. Discontinuation rates were similar between the 2 treatment groups, with common reasons for discontinuing study agent including withdrawal of consent and AE.
The safety events through 1 year of the GO-ALIVE trial were consistent with other anti-TNF therapies in AS; although it should be noted that this study was not powered to detect rare events. Infections were the most common type of AE, and the rates of infections were consistent with those in other populations managed with biologic agents16. Eight SAE occurred in GOL-treated patients; all but 2 (pneumonia and pulmonary tuberculosis) were considered by the investigators to be unrelated to study treatment. There were 3 serious infections among GOL-treated patients (pneumonia and pulmonary tuberculosis mentioned above, and appendicitis). The event of pulmonary tuberculosis was a new infection, not a reactivation, in a country where tuberculosis is more common than in Western Europe and North America. No opportunistic infections, demyelinating events, malignancies, or deaths occurred during the study. Infusion reactions were uncommon, and none were considered serious or severe or led to study discontinuation.
The totality of the results of the GO-ALIVE study demonstrated that treatment with IV GOL 2 mg/kg at weeks 0, 4, and every 8 weeks thereafter was effective in reducing the signs and symptoms of AS among adult patients with active disease despite treatment with NSAID, with sustained response through 1 year.
Acknowledgment
The authors thank Stephen Xu, MS, of Janssen Research & Development LLC, for statistical support, and Rebecca Clemente, PhD, of Janssen Scientific Affairs LLC, for writing support.
Footnotes
This study was funded by Janssen Research & Development LLC, which manufactures golimumab.
Drs. Caldron and Dudek served as trial investigators for Janssen. Dr. Reveille has received consultancies from Eli Lilly, Janssen, Novartis, Pfizer, and UCB, has served as a trial investigator for Janssen and Eli Lilly, and has received a research grant from Janssen. Dr. Deodhar has received consultancies, speaking fees, and/or honoraria from Eli Lilly, Janssen, Novartis, Pfizer, and UCB. Drs. Harrison and Hsia, and L. Kim, K.H. Lo, and J.H. Leu are or were employees of Janssen Research & Development LLC at the time this work was performed, and own stock in Johnson & Johnson, of which Janssen Research & Development LLC is a wholly owned subsidiary.
Full Release Article. For details see Reprints and Permissions at jrheum.org
- Accepted for publication November 22, 2018.
Free online via JRheum Full Release option








{kind=link}
{kind=link}