

Abstract
Objective. We aimed to jointly investigate the role of antipeptidyl arginine deiminase type 4 antibodies (anti-PAD4) and polymorphisms in the PADI4 gene together with clinical variables in rheumatoid arthritis (RA).
Methods. Serum IgG autoantibodies to human recombinant PAD4 were identified by DELFIA technique in 745 patients with RA (366 available from previous studies). Genotyping of PADI4 was performed using TaqMan assays in 945 patients and 1118 controls. Clinical data, anticitrullinated protein antibodies (ACPA) status, shared epitope status, and a combined genetic risk score were also available.
Results. Anti-PAD4 antibodies were detected in 193 (26%) of 745 patients with RA; 149 (77%) of these were also ACPA-positive. No association was observed between anti-PAD4 status and clinical characteristics, PADI4 polymorphisms, or genetic risk scores after stratification for ACPA status.
Conclusion. Taken together, the results from these combined serological, genetic, and clinical analyses suggest that anti-PAD4 appears to be a bystander autoantibody with no current clinical utility in RA.
Rheumatoid arthritis (RA) is a chronic multifactorial immune-mediated disease of unknown etiology causing cartilage and bone destruction in association with distinct serum autoantibodies1,2. The genetic component is estimated to account for 60%1, and the major genetic determinant is certain alleles in the HLA–DRB1 gene encoding the shared epitope (SE)3,4, whereas smoking is the strongest environmental risk factor identified5. The major autoantibodies in RA are anticitrullinated protein antibodies (ACPA). The ACPA recognize protein-bound citrulline residues generated posttranslationally by a family of enzymes called the peptidyl arginine deiminases (PADI). Both PADI2 and PADI4 have been shown to be associated with RA6,7,8,9,10. PADI4 has been genetically associated with RA, and it is a target of anti-PAD4 autoantibodies.
PADI4 is expressed in T cells, B cells, macrophages, neutrophils, fibroblast-like cells, and endothelial cells in the lining and sublining areas of the RA synovia11. Studies have previously reported anti-PAD4 antibodies to be associated with a more aggressive disease course12,13,14,15. Moreover, PADI4 deficiency reduced severity in an arthritis mouse model16. Anti-PAD4 antibodies have been found in sera from patients with preclinical RA (i.e., several years prior to diagnosis), but only in one-third of patients was anti-PAD4 detected before ACPA17.
The PADI4 gene, tagged by rs2301888, is among the 101 RA susceptibility loci reported in 2014 in a large genome-wide association study (GWAS) comprising 100,000 individuals18. In 2003, the PADI4 gene had already been found to be associated to both disease susceptibility and severity in Asian populations19,20. The association was, however, less clear in white populations21,22,23,24, and a metaanalysis concluded that the PADI4 polymorphism, rs2240340, was associated with RA in Asians but not in Europeans25. The RA association seen in Japan was with a functional PADI4 haplotype influencing gene expression levels, and the susceptibility haplotype was associated with a more stable mRNA (PADI4 mRNA), and therefore potentially higher levels of PADI4 proteins19.
Not until 2012 was the PADI4 gene convincingly associated with risk of RA in whites, by rs2040336, a polymorphism in weak linkage disequilibrium (LD) to those detected in Asians26. The discrepant observations regarding involvement of PADI4 polymorphisms could be due to differential LD patterns between Asian and white populations. Another explanation could be that the PADI4 variants do not exert risk for all patients with RA, but only for certain clinical subphenotypes (e.g., according to severity or antibody status). PADI4 polymorphisms (represented by rs1748033) have previously been reported to predispose male smokers for RA27, and the genetic heterogeneity between Asian and European populations was suggested to be explained by differences in smoking prevalence among men27. Additionally, the influence of anti-PAD4 on genetic profiles has not yet been addressed.
In this study, we aimed to jointly investigate the role of PADI4 polymorphisms, anti-PAD4, and clinical variables in RA. Our cohort is the largest that has been characterized for anti-PAD4 status so far. In addition, we have targeted polymorphisms with diverging LD patterns between Asian and white populations to address the population discrepancies seen for PADI4 polymorphisms and RA association.
MATERIALS AND METHODS
Patients and controls
The RA cohort comprises 945 cases diagnosed according to the 1987 American College of Rheumatology criteria28. The patients were recruited from 3 RA cohorts and 1 register study: the European Research on Incapacitating Disease and Social Support (EURIDISS) cohort, a cohort of patients with early RA undergoing magnetic resonance imaging, a cohort of patients starting tumor necrosis factor inhibitor therapy, and the Oslo RA register (ORAR)13,29,30,31. Data on ACPA and rheumatoid factor (RF) status were available for 886 of the 945 patients.
Most RA cases (n = 745) had serum samples available for anti-PAD4 detection. Of these, 366 patients had previously been assessed for anti-PAD412. For anti-PAD4 delineation on ELISA assay, 70 healthy controls (matched for age, sex, and county of residence) were used.
Analyses investigating whether disease activity varied according to anti-PAD4 antibody status were restricted to patients from the ORAR, because this represents the largest patient population (n = 395) and the inclusion criteria and clinical information available varied between cohorts. These variables were obtained from examinations performed in 1996/97 of patients included in the ORAR: 28-joint count Disease Activity Score (DAS28), 28-joint swollen joint count (SJC28), 28-joint tender joint count (TJC28), joint deformity count, C-reactive protein (CRP), erythrocyte sedimentation rate (ESR), and modified Health Assessment Questionnaire (mHAQ).
All 945 patients and 1118 controls from the Norwegian Bone Marrow Donor Registry had DNA available and were included in genotyping of PADI4 single-nucleotide polymorphisms (SNP). Genotyping data for HLA-DRB1 and SE information were available for patients and controls32, as were the genetic risk score (GRS) based on HLA-DRB1 and 22 non-HLA–risk SNP33.
The regional ethics committee approved this study (approval numbers: 281/98, 37/96, 764-04172, S-04279), and all participants gave written informed consent.
Anti-PAD4 antibody assay
The full-length human recombinant PADI4 cDNA was provided by Dr. M. Yamada (Graduate School of Integrated Science, Yokohama City University, Yokohama, Japan), expressed as a fusion protein with the 26 kDa glutathione S-transferase and purified as previously described34. The detection of IgG autoantibodies to human recombinant PAD4 (hPAD4) in RA sera was done by a dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA) technique as described elsewhere12. Briefly, active hPAD4 (5 µg/ml) were incubated overnight, followed by blocking and adding of 100 μl serum (diluted 1:2000). For detection, rabbit anti-human IgG (1:10000), biotinylated goat antirabbit IgG (1:8000), and Europium-labeled streptavidin were used and the signals were detected (Wallac). Each sample was analyzed in triplicate and 4 healthy control samples were included on each plate. Anti-PAD4 responses were calculated by subtracting the mean background signal (wells without serum) from the mean anti-PAD4 signal, and samples having a signal above the 95% percentile of the signal from the healthy control samples (n = 70) were assigned as anti-PAD4 positive.
SNP selection and TaqMan genotyping
Genomic DNA was extracted manually from blood samples by using a salting out method, and whole genome amplification by RepliG (Qiagen). The selection of SNP was based on previous associations, and on the discrepancies between the PADI4 associations observed in Asians [rs2240340 (intron SNP)] versus whites. We hypothesized whether the lack of association seen in whites (as opposed to Japanese) with rs2240340 was due to difference in LD patterns, and therefore selected candidate SNP (minor allele frequency > 10%) that were in LD with rs2240340 in the Japanese population (r2 > 0.8) but not in LD in whites (r2 < 0.8). We used HapMap release 22, Chr 1: 17507279-1756308, and identified 22 SNP fulfilling these criteria (shown in Supplementary Table 1, available with the online version of this article), which were tagged by the 4 SNP rs10788664, rs1635579, rs2240335, and rs755449 (r2 > 0.9 in the white population).
Additionally, rs2240336 within the PADI4 locus (intron SNP) was selected from the RA immunochip study26, and this SNP is in LD (r2 = 0.83) with the PADI4 SNP (rs2301888) identified in the recent and largest GWAS18. The 6 SNP were genotyped using allele discrimination TaqMan assays (Applied Biosystems): C_16176717_10 (rs2240340), C_7541127_10 (rs1635579), C_1296956_10 (rs10788664), C_3123006_1 (rs2240335), C_3123007_1_ (rs2240336), and C_937989_10 (rs766449). Only SNP with a genotype success rate > 95% and in Hardy-Weinberg equilibrium (HWE) in both patients and controls (p > 0.01) were included for analyses. All SNP had genotype success rates > 97%, but rs766449 deviated from HWE (p = 0.0006) and was excluded from further analyses. Given that the allele frequencies are > 30% and published OR > 1.2, we have > 80% power to detect an association (p < 0.05) with RA in our dataset [calculated using PS power and sample size calculation (ps-power-and-sample-size-calculation.software.informer.com)].
Calculating GRS
GRS were calculated as standardized GRS, as described by Maehlen, et al33. GRS-22 contained genetic data on 22 non-HLA RA associated loci, whereas GRS-31 consisted of the 22 genetic loci plus all 8 SE alleles, in addition to the protective effect of DRB1*13:01. Mean GRS was 0 in controls, meaning no increased genetic risk to develop RA.
Statistics
Continuous clinical variables were tested for association with anti-PAD4± stratified for ACPA status using 1-way ANOVA or Mann-Whitney U test for continuous data (age at onset, disease duration, CRP, ESR, mHAQ, DAS, SJC28, and TJC28) or using Pearson’s chi-square test for dichotomous variables (sex, RF status, smoking, SE status, and total deformed joints). P values < 0.004 were considered significant after correction for multiple testing (n = 14, using Bonferroni correction). We performed univariate and multivariate linear regression models to test whether anti-PAD4 was associated with disease outcome. In the multivariate regression, we corrected for factors known to influence the disease outcome variables of RA: ACPA, sex, age at disease onset, and disease duration. Residual plots were inspected for all outcomes in both the univariate and multivariate regression analyses. Clinical characteristics were compared in R v.3.3.3 (2017-03-06; R Foundation for Statistical Computing). Plink v1.07 was used to perform chi-square allelic tests to investigate the associations between SNP and phenotypes (pngu.mgh.harvard.edu/purcell/plink). LD was calculated based on 99 HapMap individuals (Utah residents with Northern and Western European ancestry from the Centre d’Etude du Polymorphisme Humain collection; CEU) and downloaded from 1000 Genomes35. An LD map with gene structure was generated in Haploview v4.236. The greyscale of the LD squares represents their r2 values, ranging from r2 = 0 (white) to r2 = 1 (black). Differences between the standardized GRS means were calculated using 1-way ANOVA (using built-in general statistics in R Foundation). For graphic presentations, R: a language and environment for statistical computing (R Foundation) was used.
RESULTS
Clinical characteristics of patients according to anti-PAD4 status
Among the 379 patients with RA presently tested for PAD4 antibodies, the proportion of anti-PAD4 positive was 27.7% (n = 105). The proportion of anti-PAD4–positive patients was 25.9% in our total RA cohort (n = 745), when combining 366 patients previously tested for anti-PAD412 with our 379 presently anti-PAD4–tested patients with RA.
The demographics of all the 745 patients with RA stratified for anti-PAD4 status are shown in Supplementary Table 2, available with the online version of this article. Differences between anti-PAD4–positive and -negative were seen in female sex (82.9% vs 75.9%, p = 0.04), ever smoker status (54.3% vs 66.6%, p = 0.004), RF positivity (64.0% vs 49.9%, p = 0.001), and in particular, ACPA positivity (77.2% vs 57.4%, p = 1.0 × 10−6).
The distribution of patients with RA according to both ACPA and anti-PAD4 status is shown in Figure 1. The majority of the patient cohort was positive for ACPA but negative for anti-PAD4 (42%). Further, 20% of patients were positive for both ACPA and anti-PAD4, while only 6% patients were anti-PAD4–positive and ACPA-negative.

The proportions (%) of patients with rheumatoid arthritis (n = 745) according to antibody status for anti-PAD4 and ACPA. ACPA: anticitrullinated protein antibodies; aPAD4: antipeptidyl arginine deiminase type 4.
ACPA status may influence the patient characteristics, and after stratification for ACPA status, most differences between anti-PAD4–positive and -negative patients disappeared (Table 1). Among ACPA+ patients, the proportion of ever smokers was lower in anti-PAD4+ vs anti-PAD4− patients (54.5% vs 69.0%, p = 0.004); this difference was not seen in ACPA− patients. In ACPA− patients, there was a higher percentage of females in anti-PAD4–positive patients compared with anti-PAD4–negative patients, though this difference was only borderline significant (88.6% vs 74.5%, p = 0.041). None of these associations withstood correction for multiple testing (significance threshold p < 0.004).
Demographics of the patients with rheumatoid arthritis (n = 745) after stratification for antibody status (ACPA and anti-PAD4).
To examine whether anti-PADI4 influence disease activity, we investigated the ORAR patients (n = 395). Their clinical characteristics after stratification for anti-PAD4 are shown in Supplementary Table 3, available with the online version of this article. The anti-PAD4–positive patients had a higher DAS28 score (4.7 vs 4.35, p = 0.043), SJC28 (8.0 vs 6.0, p = 0.02), and CRP (11.5 vs 9.0, p = 0.006), but also displayed a longer disease duration (14.3 vs 11.7, p = 0.012) and a larger proportion of ACPA and RF positivity (p < 0.02). Next, we examined whether anti-PAD4 could predict clinical outcomes by using a linear regression model. The univariate model (Table 2) supported the association with DAS28 (p = 0.04), SJC28 (p = 0.005), and CRP (p = 0.008). The multivariate model (Table 2) demonstrated no effect of anti-PAD4 antibody status on any of the clinical variables after adjusting for ACPA, sex, age at onset, and disease duration. These findings are in accordance with the cumulative probability plot (Supplementary Figure 1, available with the online version of this article), where anti-PAD4 status does not seem to influence the DAS28 scores (p > 0.2). In contrast, ACPA+ patients had on average, as expected, a higher DAS28 than ACPA− patients.
The influence of anti-PAD4 on clinical outcome in patients with rheumatoid arthritis participating in the ORAR registry study (n = 395).
Association analyses of PADI4 polymorphisms according to antibody status, sex, and smoking status
The distribution of genotypes and alleles of the 5 PADI4 SNP did not differ significantly (p > 0.1) between patients with RA (n = 945) and controls (n = 1118; Supplementary Table 4, available with the online version of this article). Because the genetic profile of patients with RA is known to differ based on autoantibody status33, we first stratified according to ACPA. In ACPA− patients versus controls (Supplementary Table 5), rs2240340 and rs1635579 showed a weakly significant association with RA (p = 0.045 and p = 0.016, respectively), while no association was seen in ACPA+ patients (Supplementary Table 6). When stratifying for both anti-PAD4 and ACPA (Figure 2A), the 2 SNP (rs2240340 and rs1635579) appeared to be associated only with RA in patients lacking both ACPA and anti-PAD4 (p = 0.024). This association (Pc = 0.48) did not withstand correction for multiple testing. None of the SNP showed any allelic association with RA when stratifying only for anti-PAD4 status (data not shown). The LD values between the PADI4 SNP are displayed in Figure 2B.

A. Association of the 5 tested PADI4 polymorphisms with RA stratified for autoantibody status. X axis shows the SNP base pair position on chromosome 1. Y axis shows the log-transformed p values. Dots represent the p value for corresponding SNP subgrouped into antibody subgroups of RA as well as for all patients with RA. B. LD (r2) between PADI4 polymorphisms tested in this study and previously reported to be associated with RA (based on CEU samples from the 1000 genomes project). RA: rheumatoid arthritis; ACPA: anticitrullinated protein antibodies; aPAD4: antipeptidyl arginine deiminase type 4; CEU: Utah residents with Northern and Western European ancestry from the Centre d’Etude du Polymorphisme Humain collection, HapMap; SNP: single nucleotide polymorphism; LD: linkage disequilibrium.
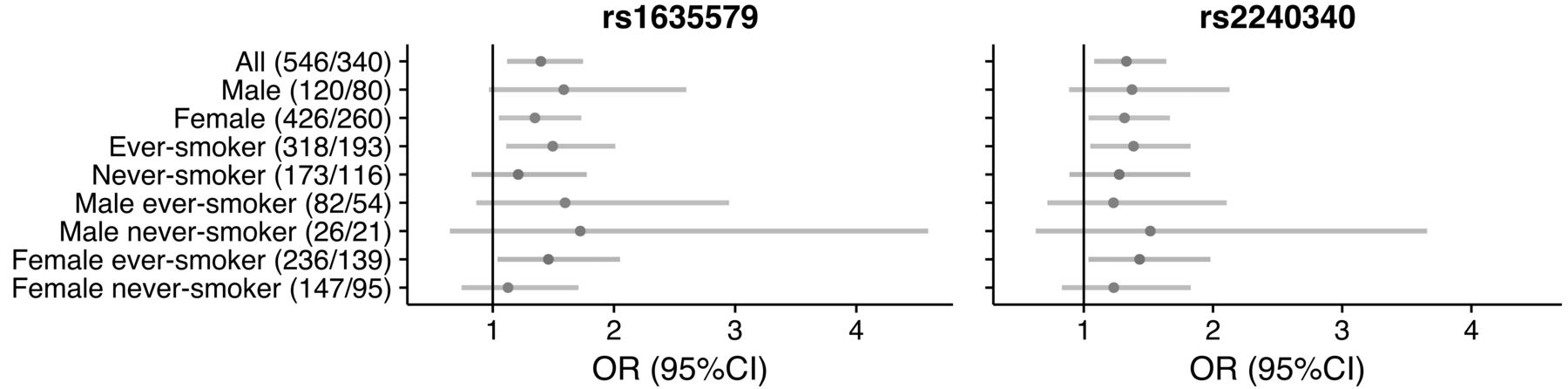
In light of the association seen between PADI4 polymorphisms (particularly rs1748033) and male smokers in RA27, we examined the association for the PADI4 polymorphisms rs2240340 and rs1635579 (r2 = 1 between rs1635579 and rs1748033, Figure 2B) in different strata according to sex, smoking, and ACPA status. A weak association was seen in ever smokers when comparing ACPA-positive and -negative patients; however, no association was observed in the strata of male smokers in particular (Figure 3, and Supplementary Table 7, available with the online version of this article).

OR with 95% CI for ACPA+ vs ACPA− when carrying the minor allele at rs1635579 or rs2240340. Numbers of patients in the different subgroups are shown in brackets (no. ACPA+/no. ACPA−). ACPA: anticitrullinated protein antibodies.
Genetic risk score and autoantibodies (ACPA and anti-PAD4)
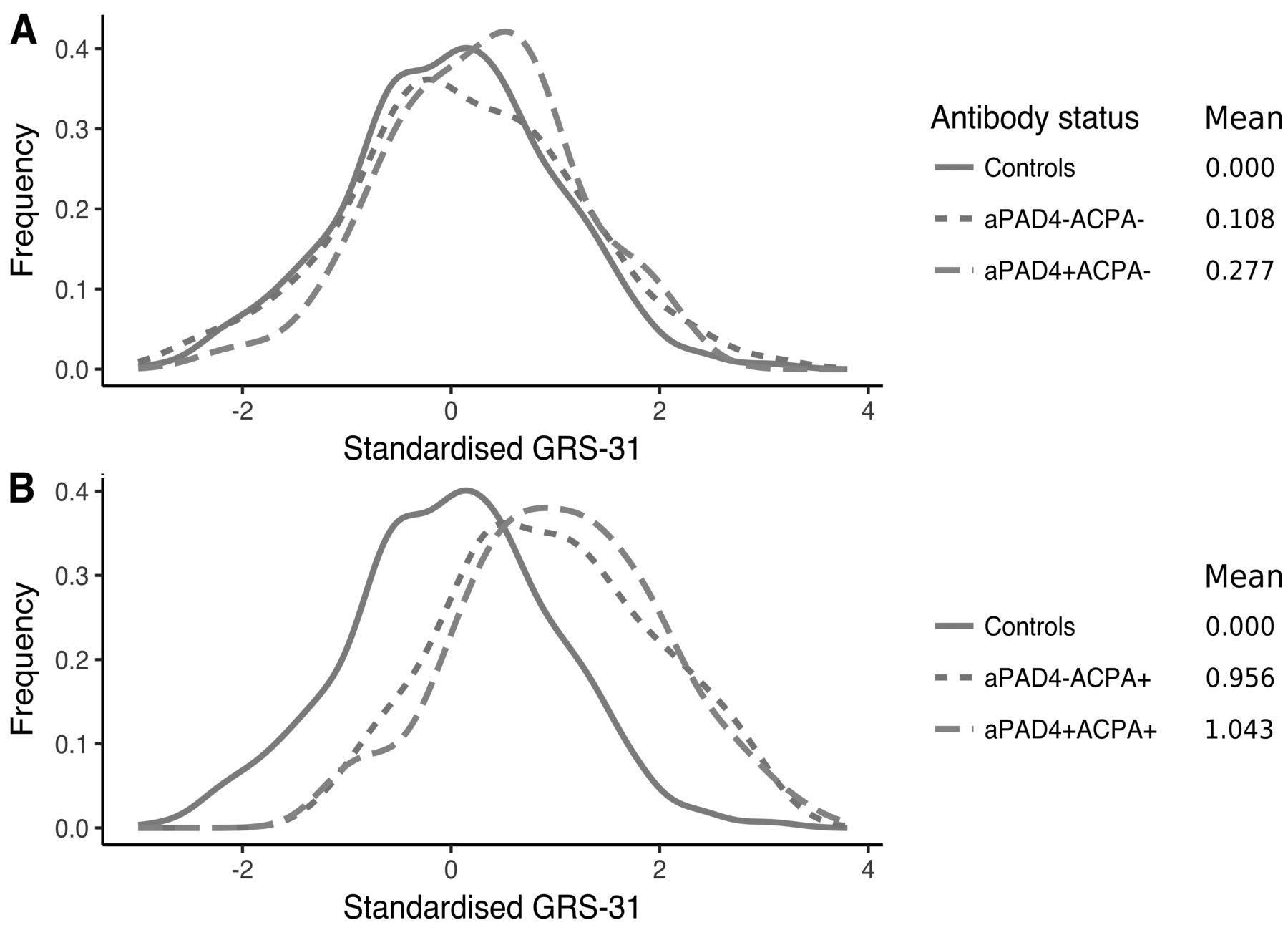
We wanted to investigate whether a more complex genetic risk profile differed between antibody subgroups. There was no association between cumulated genetic risk scores (GRS-22 and GRS-31 risk scores) and anti-PAD4 status (Supplementary Figure 2, available with the online version of this article, and Figure 4). In GRS-31, single anti-PAD4+ patients did not show increased genetic risk compared to double antibody–negative patients (anti-PAD4−ACPA−; mean GRS-31: 0.28 vs 0.11, p = 0.34, Figure 4A). In contrast, anti-PAD4−ACPA+ patients had higher GRS-31 compared to anti-PAD4−ACPA− patients (mean GRS-31: 1.04 vs 0.11, p = < 2*10−16), implying that GRS-31 is associated with ACPA status, not anti-PAD4 status. Finally, the double-positive group (anti-PAD4+ACPA+) did not have an increased risk compared to single ACPA+ patients (mean GRS-31: 1.04 vs 0.96, p = 0.38; Figure 4B). In addition, HLA-DRB1 alleles were equally distributed between anti-PAD4−positive and -negative patients (data not shown).
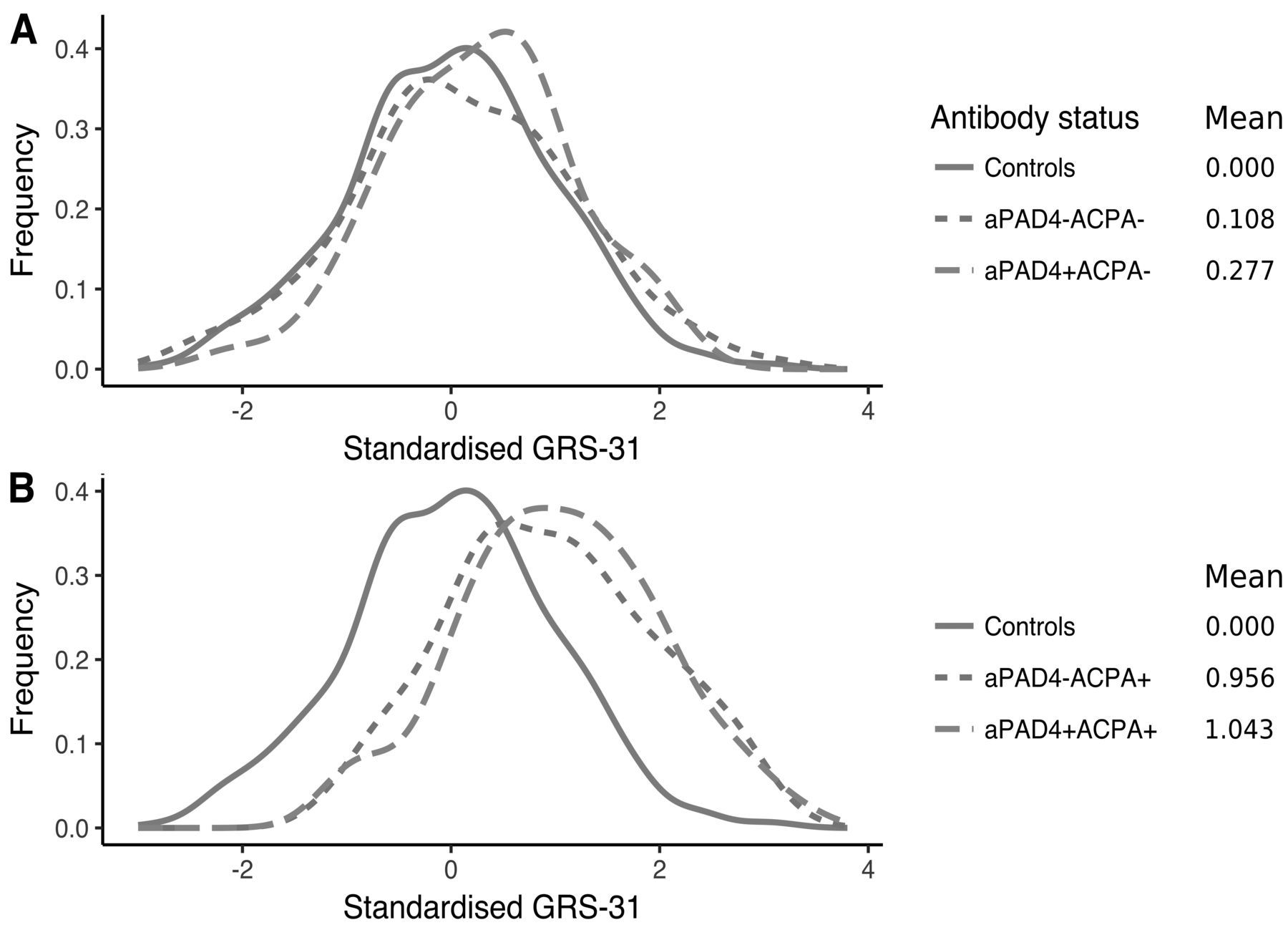
Distribution of standardized GRS-31 in controls (n = 1121) and patients (n = 744) stratified for anti-PAD4 and ACPA status. A. Controls and ACPA− patients stratified for anti-PAD4; aPAD4− ACPA− (n = 235) and aPAD4+ACPA− (n = 44). B. Controls and ACPA+ patients stratified for anti-PAD4; aPAD4− ACPA+ (n = 316) and aPAD4+ACPA+ (n = 149). ACPA: anticitrullinated protein antibodies; aPAD4: antipeptidyl arginine deiminase type 4; GRS: genetic risk score.
DISCUSSION
The frequency of anti-PAD4 varies among studies, and occurs in 22–50% of patients with RA12,14,37,38,39. An increase in anti-PAD4 positivity by disease duration has been proposed14,38 and may explain the differences in observed anti-PAD4 frequencies14, as well as use of assays directed also against citrullinated residues due to autocitrullination40. The Norwegian patients analyzed for anti-PAD4 in our study had similar frequency (27.7%) to the previously tested cohort (23%) from Norway12. The frequency observed is in accordance with the low sensitivity reported for this antibody17, and anti-PAD4 status also differed with disease duration in our ORAR patients. In addition, anti-PAD4 does not appear to be disease-specific, because it has been detected in other rheumatic diseases, although with lower titers12,15.
In contrast to what had been reported in other studies, we did not observe any significant association between disease severity (represented by DAS28, SJC, ESR) and anti-PAD4 status in the multivariate analyses14,41,42. Although this is a relatively large study (n = 395), there are 2 important limitations: first, the lack of radiographic data and second, the cross-sectional design. Radiographic damage reveals longterm inflammation while DAS28 reflects current disease activity; therefore, a longitudinal study with radiographic data would better address the question of whether anti-PAD4 influence disease severity. An association with radiographic damage has previously been reported in a subset of our cohort with longitudinal data (EURIDISS, n = 177)12. Interestingly, Darrah, et al found that a subset of anti-PAD4 positive patients with cross-reactive antibodies to PAD3/PAD4 had a higher baseline radiographic score and higher likelihood of radiographic progression, a finding later replicated41,42. Similarly, presence of anti-PAD3/PAD4 antibodies was associated with interstitial lung disease in patients with RA, another marker of disease severity43. Moreover, anti-PAD3/PAD4 antibody increased the efficiency of PADI4 by reducing the enzyme’s calcium requirement, which therefore highlights a positive feed forward loop that may (partly) explain the association between anti-PAD4 and disease severity. Unfortunately, we do not have data available on anti-PAD3/PAD4 antibodies in our cohort and thus cannot explore this hypothesis.
Our lack of association between anti-PAD4 and clinical variables are in accordance with previous observations that anti-PAD4 is mainly detected in established RA39 and suggests that the production of anti-PAD4 antibodies appears to be driven by processes that are not directly linked to the generation of ACPA antibodies. It has been hypothesized that ACPA develops first against proteins that have been citrullinated by the PADI4 enzyme, and that PADI4 later becomes an antigen44. It is also suggested that the association between ACPA and anti-PAD4 could be due to epitope spreading. Interestingly, antibodies to bacterial PAD from Porphyromonas gingivalis (PPAD), and citrullinated epitopes from PPAD, are also found in RA, but in a similar way to anti-PAD4, these antibodies also develop after the ACPA response45,46. Intriguingly, we observed the presence of anti-PAD4 in the absence of ACPA in 6% of the patients. This is in line with the observations of anti-PAD4 in other autoimmune diseases not characterized by ACPA. However, it may also be possible that these PAD-positive−ACPA-negative patients have antibodies against citrullinated proteins not detected by ordinary anticyclic citrullinated peptide assay47.
Regarding smoking, we found that there was a lower proportion of smokers in anti-PAD4+ patients than anti-PAD4− patients in the ACPA-positive subset. One could hypothesize whether epitope spreading, preceding clinical onset of RA, is facilitated by either smoking and/or PAD4 activation, because this could explain the lower incidence of smokers seen in the anti-PAD4+ patients.
No association was observed between anti-PAD4 status and PADI4 SNP in our cohort (n = 745). This has been investigated only in 1 previous study of a Mexican RA cohort (n = 170), and their findings are in accordance with ours38. Further analyses in subgroups stratified for both ACPA and anti-PAD4 status showed a borderline association in ACPA-negative, anti-PAD4–negative patients having the minor allele of rs2240340 and rs1635579. There is solid evidence that genetic risks are highly ACPA-dependent48. Hence it is crucial to stratify patients by ACPA status, although smaller patient groups increase the chance of type 2 errors and reduce power. This association did not withstand correction for multiple testing and could be a false-positive finding.
The failure to detect association to PADI4 earlier in whites could be explained by population differences in LD patterns, and rs2240340 might just be a tag SNP for the causal variant only in Asians. Alternatively, the predisposition conferred by PADI4 polymorphisms could be restricted to certain RA subphenotypes. We did not find any signs of association restricted to specific RA strata, such as autoantibody status, or between male smokers and PADI4 SNP. The latter has previously been suggested to explain the genetic PADI4 heterogeneity with RA susceptibility between Asians and Europeans, given differences in smoking prevalence27. Interestingly, rs2301888, which has recently been revealed to be associated with RA in both Asian and white populations, is in strong LD with the initial PADI4 SNP, rs2240340, associated in only Asian and not white populations. Hence, the delayed detection of an association with PADI4 in whites seems to be explained by differential LD patterns.
No association was shown between anti-PAD4 status and either PADI4 SNP, SE, HLA-DRB1 alleles, or a more complex genetic risk score. ACPA alone showed a higher genetic RA risk profile compared to ACPA-negative patients or controls. It has previously been demonstrated that a higher risk of RA exists with an increased number of antibodies (RF and ACPA)33, but this additional risk is not seen with anti-PAD and ACPA. A limitation of the tested genetic risk factors is that most genetic studies in RA have been performed in the ACPA+ patient groups, and other genetic variants could be of importance in anti-PAD4 development.
Anti-PAD4 detection does not provide any clear clinical value, especially when compared with ACPA. Whether anti-PAD4 has a prognostic value regarding radiographic disease progression should be addressed in additional studies. The lack of association between anti-PAD4, clinical characteristics, and genetic risk factors suggests that anti-PAD4 is not a primary driver of the RA pathogenesis.
ONLINE SUPPLEMENT
Supplementary material accompanies the online version of this article.
Acknowledgment
The Norwegian Bone Marrow Donor Registry is acknowledged for access to the healthy controls. We thank Michiyuki Yamada for the hPAD4 construct.
Footnotes
This project is financed by the Southern and Eastern Heath Region of Norway (Helse Sør-Øst).
- Accepted for publication February 6, 2018.








{kind=link}
{kind=link}
{kind=link}
{kind=link}