

To the Editor:
A 40-year-old woman was diagnosed with systemic lupus erythematosus (SLE) 10 years ago, manifesting with edematous hands, Raynaud phenomenon, livedo reticularis, arthralgias/arthritis of hand joints, and lymphadenopathy, without renal involvement. Laboratory and serological findings included chronic disease anemia, leukopenia, and diffuse hypergammaglobulinemia; positive antinuclear, anti-Sm, anti-Ro (Ro52 and Ro60), and increased anti-dsDNA antibodies; and hypocomplementemia. For the last 2 years, she was in remission under methylprednisolone 4 mg/day, azathioprine 100 mg/day, and hydroxychloroquine 200 mg/day. Two months before presentation, feeling inundated by the chronicity of her disease and treatment, she discontinued all medication. Upon presentation, she was unwell and dyspneic, and had intense livedo of lower extremities, Raynaud phenomenon, edematous hands with arthritis, and a distended abdomen. Breathing sounds of the left lung base were reduced. There was no pericardial friction rub or pulsus paradoxus, and echocardiography revealed a nonhemodynamically relevant pericardial effusion. Chest and abdominal computed tomography showed, in addition to the pericardial effusion, a unilateral pleural effusion (Figure 1A) and massive ascites (Figure 1B). Remarkable laboratory tests included elevated C-reactive protein [CRP; 30 mg/l, normal value (nv) < 5 mg/l], diffuse hypergammaglobulinemia (gamma-globulins 34%), marked hypocomplementemia (C3 31 mg/dl, C4 < 3 mg/dl), and increased serum CA-125 (85 IU/ml, nv < 30 IU/l). Ascitic and pleural fluid analyses showed exudate effusions (ascites-protein/serum-protein = 0.6 and pleural effusion-protein/serum-protein = 0.6) and serum-ascites albumin gradient (1.1 g/dl). Multiple cultures and cytospin of both effusions were unrevealing. Considering the presence of ascites, pleural effusion, and elevated CA-125 levels, and after excluding underlying malignancy by imaging of the thorax and abdomen, and by cytological examination of the effusions, the diagnosis of pseudo-pseudo Meigs syndrome (PPMS) in the context of SLE exacerbation was made. The patient was given intravenous methylprednisolone pulses (1 g/d for 3 consecutive days), followed by mycophenolate mofetil (MMF; 3 g/d) and oral methylprednisolone (16 mg/d). Two months later, after initial temporary improvement, the patient (while still receiving the same treatment) presented again to our outpatient clinic with increasing dyspnea and abdominal distension. Complete clinical, laboratory, and imaging reevaluation again revealed exudative effusions, with laboratory and serological testing indicative of SLE exacerbation. Many autoantibodies were detected in the patient’s sera, including anti-Ro, anti-Sm, anti-UnRNP, antinucleosome, antihistones, and antiriboprotein-P. MMF was discontinued and the patient was given monthly intravenous cyclophosphamide (0.75 g/m2 body surface area), along with monthly methylprednisolone pulses (1 g). Four months later, with the patient still receiving the same treatment, no pericardial, pleural, or ascitic effusions were detectable. Our present study was approved by the Ethical/Scientific Committee of the Laikon Hospital (no. 12/20-4-2016), and the patient provided informed consent according to the Declaration of Helsinki.
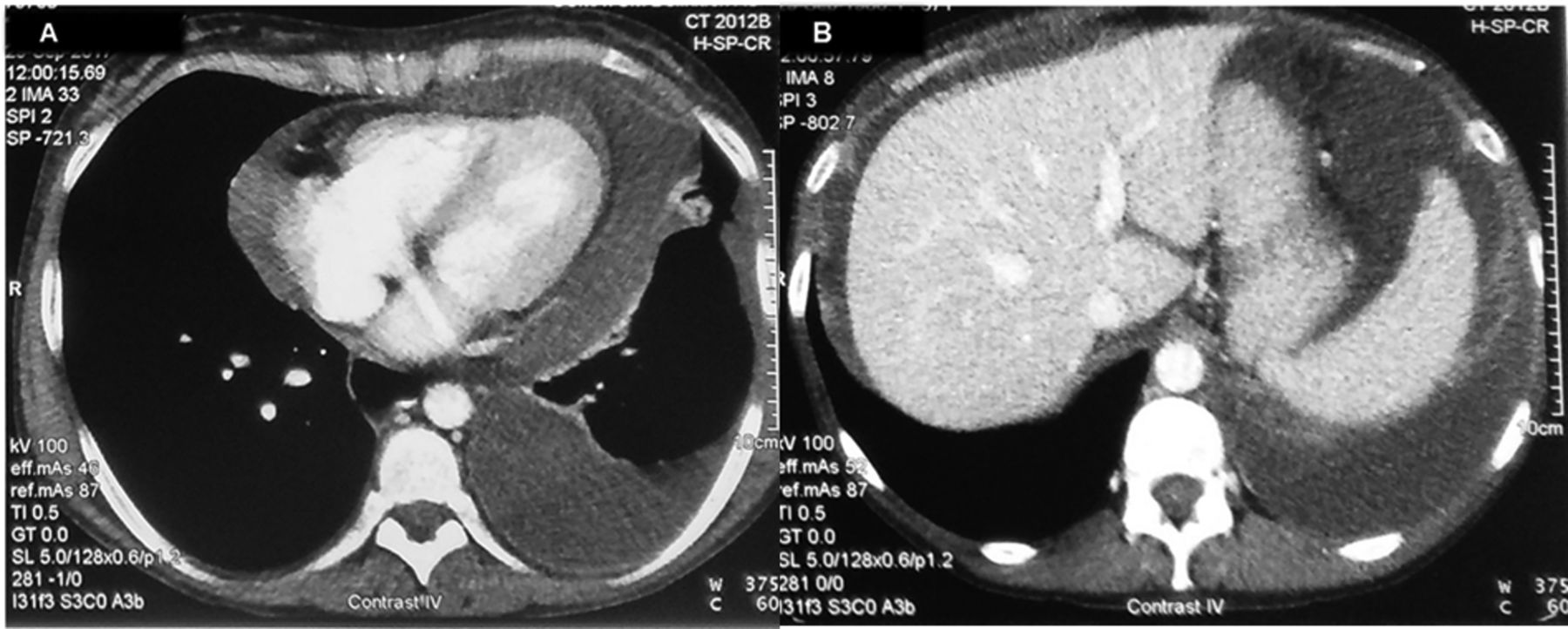
Chest and abdominal CT showed, in addition to the pericardial effusion, a unilateral pleural effusion (A) and massive ascites (B). CT: computed tomography.
Meigs syndrome is the presence of a benign ovarian tumor (fibroma or fibroma-like) in conjunction with pleural effusion and ascites1. Pseudo-Meigs syndrome is the coexistence of pleurisy and ascites secondary to nonfibroma ovarian or pelvic tumors2. What causes peritoneal and pleural effusions is still unclear, and contradictory data regarding the characteristics of effusions as transudates or exudates have been presented3. These syndromes are infrequent, yet are clinically important because although clinical manifestations suggest disseminated malignancy, symptoms subside after tumor excision, which is diagnostic for both syndromes.
PPMS is a rare clinical entity described in patients with SLE, characterized by pleurisy, ascites, and elevated CA-125 levels, similar to Meigs and pseudo-Meigs syndromes, but without association to any benign or malignant tumor4. There are few reports of PPMS in association to SLE (SLE-PPMS)5,6,7,8. All SLE-PPMS cases described refer to female patients of reproductive age, with hypocomplementemia, high anti-dsDNA titers, and exudative ascitic and pleural effusions with increased serum CA-125 as presenting manifestations of SLE. High CRP is reported only in SLE-PPMS cases with arthritis7,8.
The pathogenesis of effusion production in SLE-PPMS remains unknown. Type I interferon produces immune dysregulation in SLE, which in turn tilts T cell function toward Th cell phenotype, promoting B cell differentiation and release of the proinflammatory mediators interleukin 6 and tumor necrosis factors, contributing to tissue inflammation. CA-125 is an accepted tumor marker for ovarian cancer, yet serum concentrations can rise in various benign conditions. Peritoneal mesothelial cells express CA-125 constitutively, and synthesis is increased when mesothelial cells are activated by inflammatory cytokines9. Taking into account the aforementioned, we suggest that in flaring SLE patients with a large inflammatory load who present with PPMS, the excess of inflammatory cytokines results in increased vascular permeability and capillary leakage, which could be responsible for the production of pleural, pericardial, and ascitic effusions; the inflammatory load also results in the activation of peritoneal mesothelial cells to secrete CA-125.
We present this case for several reasons. First, SLE-PPMS is a rare clinical entity. Second, clinicians should be aware that increased CA-125 in patients with ascites and pleural and pericardial effusions needs careful interpretation and does not always indicate the presence of malignancy. Third, although it is known that SLE serositis does not cause significant effusions, in SLE-PPMS cases, massive polyserositis can be the initial manifestation either of the disease or of SLE exacerbation. Fourth, increased CRP in patients with SLE-PPMS is reported only in cases of patients with concomitant arthritis10. Finally, as in the case of our patient, aggressive combination immunosuppressive therapy might be required to assure a positive outcome in SLE-PPMS cases.








{kind=link}