

Abstract
Objective. To evaluate the safety and efficacy of pomalidomide (POM) on forced vital capacity (FVC), modified Rodnan skin score (mRSS), and gastrointestinal (GI) symptomatology over 52 weeks of treatment in patients with interstitial lung disease due to systemic sclerosis (SSc).
Methods. Twenty-three adult patients diagnosed with SSc were randomized 1:1 POM:placebo (PBO).
Results. Mean change at Week 52 from baseline in predicted FVC% −5.2 and −2.8; mRSS −2.7 and −3.7; and UCLA Scleroderma Clinical Trial Consortium Gastrointestinal Tract (SCTC GIT 2.0) score 0.1 and 0.0, with POM and PBO, respectively. Statistical significance was not achieved for any of these 3 primary endpoints at 52 weeks.
Conclusion. Because of recruitment challenges, subject enrollment was discontinued early. In an interim analysis, the study did not meet its Week 52 primary endpoints. Therefore, a decision was made to terminate all study phases. POM was generally well tolerated, with an adverse event profile consistent with the known safety and tolerability profile of POM in other diseases. Study results were neither positive nor negative because too few subjects were enrolled to make meaningful conclusions. Clinical Trials number: NCT01559129.
- SYSTEMIC SCLEROSIS
- INTERSTITIAL LUNG DISEASE
- CLINICAL TRIALS
- DISEASE ACTIVITY
- DISEASE-MODIFYING ANTIRHEUMATIC DRUGS
Lung involvement has become the leading cause of deaths directly attributable to systemic sclerosis (SSc)-associated interstitial lung disease (ILD)1. Progressive SSc-ILD is associated with inexorable deterioration in lung function with associated debility and death, resulting in 5- and 10-year mortality of 30% and 50%, respectively2.
Pomalidomide (POM) is an immunomodulatory compound, structurally similar to thalidomide. POM binds to cereblon and facilitates Ikaros and Aiolos degradation, resulting in immune-modulation of myeloid and lymphocyte cells. POM exhibits antifibrotic activity in preclinical models of dermal fibrosis3. Thalidomide has demonstrated beneficial effects in 11 patients with SSc treated in an open-label, dose-escalating, 12-week study4. Skin biopsies demonstrated changes in skin fibrosis and an increase in epidermal and dermal infiltrating CD8+ T cells with thalidomide treatment. Plasma levels of interleukin 12 (IL-12) and tumor necrosis factor-α increased, while IL-5 and IL-10 remained unchanged, suggesting that thalidomide had a pro-Th1 immunomodulatory effect. These changes were associated with beneficial, although limited, clinical effects, including decreased gastroesophageal reflux symptoms, and healing of digital ulcers. Nonclinical studies demonstrated that POM has potential antifibrotic effects in patients with SSc through the inhibition of Th2 cytokines and enhanced production of antifibrotic Th1 cytokines such as interferon-γ, granulocyte-macrophage colony-stimulating factor, and IL-24. These results suggested that POM might have therapeutic benefit as an antifibrotic agent to improve pulmonary and dermal fibrosis in SSc.
MATERIALS AND METHODS
Study design
This phase II, global, multicenter, randomized, double-blind, parallel-group study consisted of 2 treatment phases: 52 weeks of blinded treatment and a 2-year open-label extension phase, which was followed by up to a 4-year longterm followup phase. The study was conducted in accordance with the general ethical principles outlined in the Declaration of Helsinki and following protocol CC-4047-SSC-001 as approved by the Western Institutional Review Board, number 20111970. All patients provided their written informed consent before starting any study-related procedures.
Patients
Eligible patients between 18 and 80 years of age met the diagnosis of limited cutaneous SSc (lcSSc) or early diffuse cutaneous SSc (dcSSc) based on the 2013 classification criteria for SSc5, with onset of the first non-Raynaud SSc symptoms within 7 years prior to screening date. The initial protocol required the onset of the first non-Raynaud symptoms to be within 5 years prior to screening; however, owing to enrollment difficulty, the protocol was amended to increase the length of allowable disease duration. The 7-year duration inclusion criterion is in line with other SSc-ILD studies [e.g., SLS-1, abituzimab, and pirfenidone (LOTUSS) studies at clinicaltrial.gov]. Eligible patients were also required to meet at least 1 of the following pulmonary-related criteria: (1) forced vital capacity (FVC) ≥ 45% and < 70% of predicted value at screening and baseline (Visit 2), or (2) FVC ≥ 70% and ≤ 80% of predicted value at screening and baseline (Visit 2). They also needed to have a documented history of either or both (1) a ≥ 5% decrease in FVC in the 24-month period prior to baseline (based on 3 or more assessments), or (2) a high-resolution computed tomography (HRCT) fibrosis score > 20%6. To ensure enrollment of subjects with minimal FVC variability, a subject’s baseline FVC readings were required to be within 5% of their screening FVC readings. DLCO ≥ 35% and ≤ 80% of predicted value was required at screening in addition to abnormalities on HRCT consistent with SSc-ILD, including honeycombing or reticular changes with or without ground glass7. Subjects with pulmonary arterial hypertension (PAH) were permitted to enroll except for those who required PAH treatment during screening phase.
In the 52-week placebo (PBO)-controlled phase, about 88 patients were to be randomized 1:1 to PBO or POM 1 mg once daily (QD), stratified based on their type of SSc, either lcSSc or dcSSc. At Week 52, all placebo-assigned patients who completed the treatment phase and wished to enter into the open-label extension phase were transitioned to POM 1 mg QD in a blinded fashion for up to an additional 2 years; patients receiving POM 1 mg QD continued this assigned treatment. Patients were permitted to continue other supportive medications at stable doses including proton pump inhibitors, angiotensin-converting enzyme inhibitors/angiotensin receptor blockers, and cough medications to treat SSc disease-related symptoms. No other immunosuppressive therapy was permitted, except low-dose systemic corticosteroids (≤ 10 mg prednisone or equivalent/day).
Efficacy assessments
Coprimary efficacy assessments included spirometry testing of FVC and forced expiratory volume in 1 s, the total University of California, Los Angeles, Scleroderma Clinical Trial Consortium Gastrointestinal Tract (UCLA SCTC GIT) V2.0 score, and the modified Rodnan skin score (mRSS). This measure of skin thickening has been validated in dcSSc, but has been widely used in lcSSc, and used in our study for all patients. It has been shown to correlate with internal organ involvement and survival more often in patients with dcSSc than patients with lcSSc8, and was an appropriate primary endpoint in our study because the majority of patients enrolled (17/22, 77%) fell into the dcSSc category. Secondary efficacy assessments included baseline and transition dyspnea indices (BDI/TDI), pulse oximetry (SpO2), and the UCLA SCTC GIT 2.0 subscale scores (reflux, distension/bloating, fecal soilage, diarrhea, social functioning, emotional well-being, and constipation). Exploratory efficacy assessments included the Scleroderma Health Assessment Questionnaire (SHAQ). Skin punch biopsies were performed on subjects who provided additional consent.
Safety analysis
At scheduled clinic visits, safety was evaluated based on adverse events (AE), weight, vital signs, physical examination, electrocardiograph recordings, and clinical laboratory studies. An independent Data Monitoring Board was implemented for safety and clinical data monitoring as an additional feature for our study.
Statistical analysis
An estimated sample size of 88 patients was needed to yield 80% power to detect ≥ 5% difference between POM treatment and PBO, with 8% of common SD and a 25% discontinuation assumption, using a 2-sample t test with a 2-sided significance level of 0.1. Efficacy was evaluated based on the full analysis set, which consisted of all randomized patients who received at least 1 dose of study drug and who had at least 1 postbaseline assessment performed. Safety was evaluated based on the safety population, which consisted of all randomized patients who received at least 1 dose of study drug, and safety outcomes were analyzed and summarized descriptively.
RESULTS
Patients
A total of 59 patients were screened, 23 were randomized, and 22 received the study drug. Eleven patients were randomized in the POM arm and 12 in the PBO arm. One patient, who was ineligible for the study, was randomized in the POM arm, but did not receive study drug. Of these patients, 11 (50.0%) completed 52 weeks, with more PBO patients (7, 58.3%) completing treatment versus (4, 36.4%) POM patients. Eight subjects entered the open-label extension phase; none of the subjects completed the 2-year extension phase (7 patients discontinued due to study termination by the sponsor; 1 patient due to AE), and the average length of participation in this phase ranged from about 4 months to 10 months. The maximum length of treatment with POM was about 2 years.
The demographic and disease characteristics of patients at baseline were comparable across treatment groups (Table 1). The mean age was 49.8 years (range 31 to 69 yrs) in the POM arm and 44.8 years (range 25 to 68 yrs) in the PBO arm. Ninety percent of patients in the POM arm and 83.3% in the PBO arm were female. Seventy percent of patients in the POM arm and 83.3% of patients in the PBO arm were white. Mean (min, max) body mass index at screening was 26.67 kg/m2 (17.4, 49.8) and 30.64 kg/m2 (22.0, 40.9) in the POM and PBO arms, respectively. The majority of patients had dcSSc: 8 (80%) and 9 (75%) in the POM and PBO arms, respectively. Disease duration was comparable between the POM and PBO arms (4.7 vs 5.3 yrs, respectively). One patient entered the study with a diagnosis of pulmonary hypertension. The patient, who remained stable during the study, completed about 10 months of the double-blind treatment phase and was discontinued at the time of study termination by the sponsor.
Baseline demographic and clinical characteristics: full analysis set (n = 22). Values are n (%) unless otherwise specified.
Efficacy
Observed changes in all coprimary efficacy endpoints numerically (not statistically) favored PBO. In the POM and PBO arms, the mean change at Week 52 from baseline in predicted FVC% was −5.2 and −2.8, for the mRSS −2.7 and −3.7, and for the UCLA SCTC GIT 2.0 instrument total score 0.1 and 0.0, respectively9. The only mean improvement (−0.1) from baseline in the UCLA SCTC GIT 2.0 instrument total score was at Week 24 in the POM arm (Table 2). Statistical significance was not achieved for any of the 3 primary endpoints. Efficacy could not be evaluated during the longterm extension phase because of the small number of patients enrolled and the limited treatment period.
Primary endpoints of Week 24 and Week 52: full analysis set (n = 22).
For the secondary efficacy endpoints, numerical improvement in the POM arm was reported for the mean changes in the UCLA SCTC GIT 2.0 instrument subscale scores of diarrhea (0.2 and 0.3 at weeks 12 and 24, respectively) and emotional well-being (0.4, 0.3, and 0.1 at weeks 12, 24, and 52, respectively)10. The changes from baseline in dyspnea (as measured by the TDI) regarding functional impairment, magnitude of task, and magnitude of effort at weeks 12, 24, and 52 numerically favored PBO. The breathing problems visual analog scale of the SHAQ also numerically favored PBO at Week 52.
Only 5 subjects were enrolled in the optional skin biopsy substudy. Dermal thickness, collagen deposition (not shown), and myofibroblast counts were not consistently changed in these individual patients at 4 weeks or 52 weeks as compared to baseline. Decreased CD4 T cell counts, and to a lesser extent CD8 T cell counts, were noted in a subset of this sample; however, the sample number was too small for definite conclusions (Figure 1).
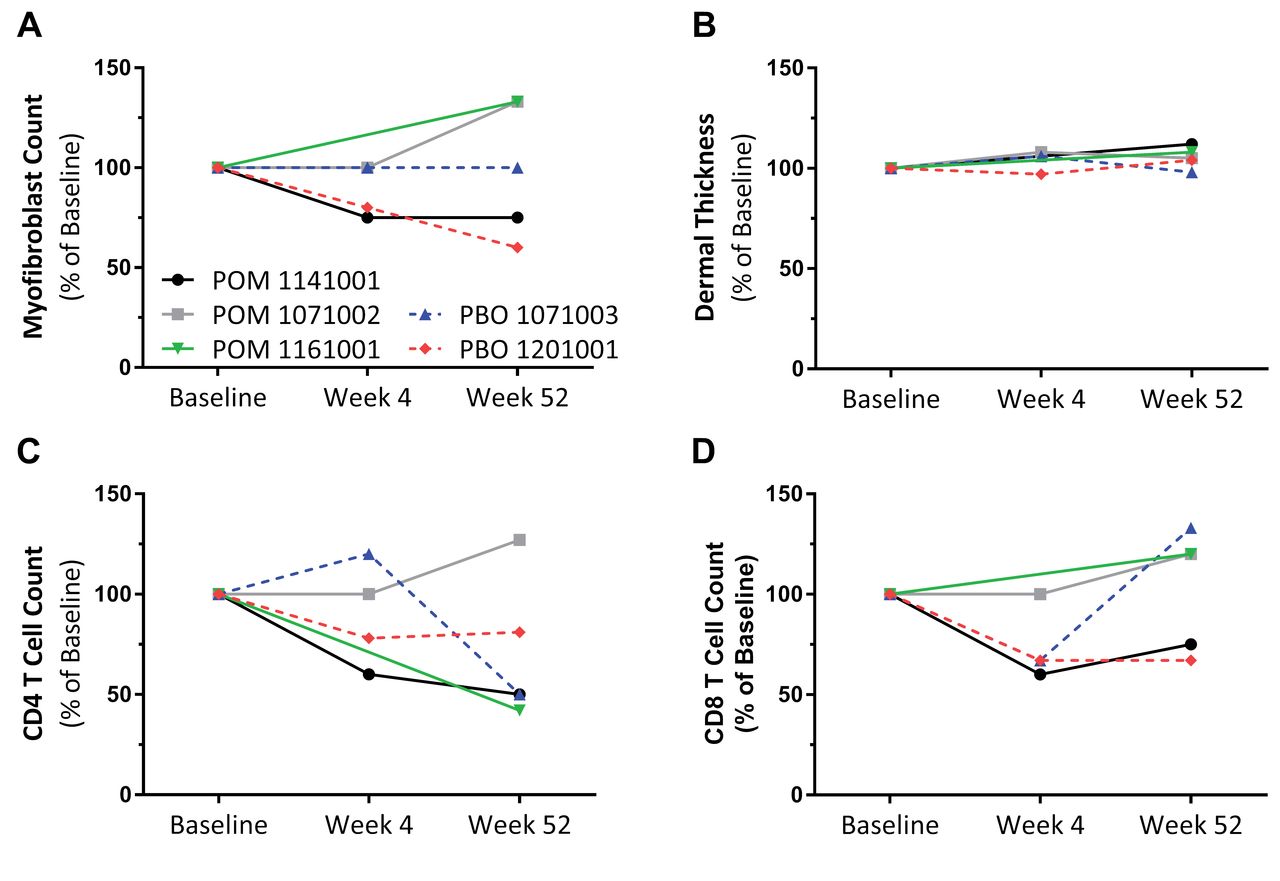
Evaluation of skin biopsies from POM- and PBO-treated subjects. Skin punch biopsies were collected at baseline, Week 4, and Week 52, and α-smooth muscle actin (myofibroblast), CD4, and CD8 immunohistochemistry was performed on sequential sections. Changes over time for individual subjects are shown as percent of baseline: (A) numbers of myofibroblasts, (B) mean dermal thickness, (C) CD4-positive cells, and (D) CD8-positive cells. POM: pomalidomide; PBO: placebo.
Safety and tolerability
In the 52-week placebo-controlled period, treatment-emergent AE (TEAE) were reported by all 12 patients receiving PBO, and 9 (90.0%) receiving POM 1 mg QD (Table 3). AE occurring in ≥ 2 patients in either treatment group during this phase included constipation, diarrhea, nausea, bronchitis, upper respiratory tract infection, influenza, urinary tract infection, ligament sprain, arthralgia, headache, dyspnea, oropharyngeal pain, rash, and skin ulcer (Table 3). In this phase, 4 POM patients (40%) discontinued owing to TEAE for the following reasons: C-reactive protein increased (associated with tendon-friction rubs, increased pain, and musculoskeletal complaints), renal failure, pulmonary embolism, and toxic skin eruption — 1 subject with each TEAE. One PBO subject reported pulmonary hypertension during this phase of the study.
Adverse events. Values are n (%) unless otherwise specified.
In the open-label extension phase, AE were reported by 7 out of 8 patients enrolled (88%); 5 of the 7 had previously been treated with placebo in the double-blind treatment phase. No patient discontinued owing to TEAE during the open-label extension phase (Table 3). There were 4 POM patients (40%) with serious AE (SAE) compared to 1 PBO patient (8.3%) during the treatment phase; 1 patient (12.5%) randomized to POM reported multiple SAE [acute respiratory failure, chronic respiratory failure, pulmonary hypertension (moderate severity)] during the open-label extension phase (Table 3). One PBO-randomized subject also reported pulmonary hypertension (not considered an SAE) following treatment discontinuation from this phase of the study.
DISCUSSION
Because of difficulties in recruiting patients for our study related to restrictive inclusion and exclusion criteria, the sponsor terminated enrollment. Based upon interim analysis data, the study did not demonstrate a statistically significant improvement in any of the 3 coprimary efficacy endpoints (changes from baseline in FVC, mRSS, or UCLA SCTC GIT 2.0 instrument total score at Week 24 or Week 52) for patients who completed blinded treatment. A limitation of this trial is that other standard patient-report outcomes such as the Medical Outcomes Study Short Form-36 were not used, which could have demonstrated different results. Patients in both the PBO and active treatment arms demonstrated disease progression consistent with the natural history of SSc-ILD. The study was terminated early because of the lack of clinical efficacy of POM over 52 weeks. At Week 52, no trends were noted for the secondary endpoints over time or by treatment group.
Our cohort was enriched with SSc-ILD patients who were at increased risk for disease progression of their pulmonary fibrosis, and because this was a placebo-controlled study, it posed great difficulty in patient recruitment. Our study was also designed to assess drug tolerability in SSc, which was unknown because most of the prior experience with this drug had been in oncology. Because there is no effective treatment for SSc-ILD, PBO-controlled trials are considered ethical11 to many investigators. However, the standard use of PBO in clinical trials often presents a challenge for recruitment. To improve recruitment, perhaps future trials in this population should consider using standard-of-care (SOC) therapy as the “placebo” arm to compare with study drug or SOC as background, with drug versus PBO as “add-on” therapy. Another option would be to offer “rescue therapy” as early as possible to those who have discontinued, which should be clearly defined in the study protocol. These patients would remain in the blinded study and be followed similarly to those who continue taking the study drug.
In our study, POM was generally well tolerated, with an AE profile consistent with the known safety and tolerability profile for POM in other diseases. There were no new safety findings from our study. More patients in the POM arm experienced serious TEAE, severe TEAE, and TEAE leading to drug withdrawal.
Although thalidomide has been shown in an open-label study to have some beneficial effects in SSc, POM did not improve the clinical measurements in this small, placebo-controlled SSc-ILD population. There were too few patients (only 4 POM subjects completed 52 weeks of treatment) to make meaningful conclusions.
Our current study did not demonstrate efficacy of POM therapy in patients with SSc-ILD. POM demonstrated an acceptable safety profile and was generally well tolerated. The safety profile was similar to previous investigations of POM for other conditions.
Acknowledgment
The authors wish to acknowledge the contributions of William Smith, MD, who served as a medical monitor for our study.
Footnotes
Funded by Celgene Corp.
- Accepted for publication August 9, 2017.








{kind=link}