

Abstract
Objective. CXCL6, a chemokine with proangiogenic property, is reported to be involved in vasculopathy associated with systemic sclerosis (SSc). We investigated the contribution of CXCL6 to SSc development by focusing on the association of friend leukemia virus integration 1 (Fli1) deficiency, a potential predisposing factor of SSc, with CXCL6 expression and clinical correlation of serum CXCL6 levels.
Methods. mRNA levels of target genes and the binding of Fli1 to the CXCL6 promoter were evaluated by quantitative reverse transcription-PCR and chromatin immunoprecipitation, respectively. Serum CXCL6 levels were determined by ELISA.
Results. FLI1 siRNA significantly enhanced CXCL6 mRNA expression in human dermal fibroblasts and human dermal microvascular endothelial cells, while Fli1 haploinsufficiency significantly suppressed CXCL6 mRNA expression in murine peritoneal macrophages stimulated with lipopolysaccharide. Supporting a critical role of Fli1 deficiency to induce SSc-like phenotypes, CXCL6 mRNA expression was higher in SSc dermal fibroblasts than in normal dermal fibroblasts. Importantly, Fli1 bound to the CXCL6 promoter in dermal fibroblasts, endothelial cells, and THP-1 cells. In patients with SSc, serum CXCL6 levels correlated positively with the severity of dermal and pulmonary fibrosis and were elevated in association with cardiac and pulmonary vascular involvement and cutaneous vascular symptoms, including Raynaud phenomenon, digital ulcers (DU)/pitting scars, and telangiectasia. Especially, serum CXCL6 levels were associated with DU/pitting scars and heart involvement by multiple regression analysis.
Conclusion. CXCL6 expression is upregulated by Fli1 deficiency in fibroblasts and endothelial cells, potentially contributing to the development of fibrosis and vasculopathy in the skin, lung, and heart of SSc.
Systemic sclerosis (SSc) is a multisystem chronic disease characterized by extensive fibrosis of the skin and various internal organs following autoimmune inflammation and vasculopathy1. Although the pathogenesis of SSc has yet to be elucidated, a series of genetic and epidemiologic studies have demonstrated that environmental factors trigger the development of this disease in individuals genetically predisposed to autoimmunity2. This notion is supported by the following evidence: (1) the biggest risk factor for SSc is family history3, (2) concordance for SSc is around 5% in twins and similar in monozygotic and dizygotic twins, whereas antinuclear antibodies are more frequently detected in the healthy monozygotic twin sibling than in the healthy dizygotic twin sibling of a patient with SSc4, (3) most of SSc susceptibility genes are HLA haplotypes and non-HLA immune-related genes which are shared by other collagen diseases5. Therefore, the identification of predisposing factors reflecting environmental influences, such as the genes regulated by an epigenetic mechanism, would be quite helpful to investigate the developmental process of SSc, as is the case with other rheumatic diseases6.
Friend leukemia virus integration 1 (Fli1) is a member of the Ets transcription factor family, the expression of which is broadly suppressed in various types of cells in involved and uninvolved skin of patients with SSc7. Fli1 expression is reduced by transforming growth factor (TGF)-β, endothelin-1, and interferon-γ (IFN-γ) in dermal fibroblasts and/or dermal microvascular endothelial cells8,9,10,11. More importantly, Fli1 expression is epigenetically suppressed in the bulk skin and cultivated dermal fibroblasts from patients with SSc12, suggesting that Fli1 deficiency serves as a predisposing factor reflecting the influence of environmental factors in this disease. Indeed, Fli1 haploinsufficiency augments the induction of SSc-like phenotypes in dermal fibroblasts, dermal microvascular endothelial cells, and macrophages in mice treated with bleomycin13. Also, Fli1 deficiency regulates the expression of various disease-related molecules in SSc14,15,16,17,18,19,20,21,22. Therefore, the molecular analysis based on Fli1 deficiency provides us with a useful clue to know the significance of target molecules in the pathogenesis of SSc.
CXCL6, also known as granulocyte chemotactic protein 2, is a member of CXC chemokines with glutamic acid-leucine-arginine (ELR) motif23. The properties of CXC chemokines generally depend on the presence or absence of the ELR motif in their N terminus; ELR+ CXC chemokines are potent promoters of angiogenesis as well as potent activators and chemoattractants for neutrophils, while ELR–chemokines are potent inhibitors of angiogenesis without neutrophil chemoattractant property24. Consistent with these unique features, CXCL6 is implicated in the development of diseases characterized by neutrophil infiltration and aberrant vascular activation. For instance, CXCL6 expression is increased in intestinal endothelial cells of patients with inflammatory bowel disease25 and in synovial fibroblasts of patients with rheumatoid arthritis26. Also, owing to its proangiogenic effect, CXCL6 serves as a marker of a highly progressive phenotype of various kinds of tumors27,28. In patients with SSc, circulating CXCL6 levels are significantly elevated, irrespective of disease subtypes [diffuse cutaneous (dcSSc) and limited cutaneous SSc (lcSSc)], and CXCL6 expression is increased in dermal microvascular endothelial cells29. Contrary to CXCL6 upregulation in endothelial cells, neutrophil infiltration is barely seen in SSc lesional skin30. Considering the upregulated expression of CXCR2, a receptor for CXCL6, and altered activation of intracellular signaling by CXCL6 in SSc endothelial cells29, CXCL6 seems to contribute to the development of SSc vasculopathy. Indeed, SSc endothelial cells are unresponsive to the migratory effect of CXCL6 in vitro29. However, the entire involvement of CXCL6 in SSc pathogenesis has remained elusive.
With this background, we here investigated the potential involvement of CXCL6 in the development of SSc. To this end, we looked at the effect of Fli1 deficiency on the expression levels of CXCL6 in dermal fibroblasts, endothelial cells, and macrophages. Also, we analyzed the association of serum CXCL6 levels with clinical features of SSc. Our results indicate a possible contribution of CXCL6 to the fibrotic and vascular aspects of SSc.
MATERIALS AND METHODS
Ethics statement
Our study was approved by the ethical committee of the University of Tokyo Graduate School of Medicine. Written informed consent was obtained from all patients and healthy individuals. The whole study was performed according to the Declaration of Helsinki. All animal studies and procedures were approved by the committee on animal experimentation of the University of Tokyo Graduate School of Medicine.
Cell cultures of fibroblasts, endothelial cells, and THP-1 cells
Human dermal fibroblasts were obtained from 5 patients with dcSSc with < 2 years of skin thickening and from the corresponding area of 5 closely matched healthy donors, and then maintained as described previously9. Human dermal microvascular endothelial cells (HDMEC), purchased from Takara Bio Inc., were cultured as described previously10. THP-1 cells, a human monocytic cell line derived from a patient with acute monocytic leukemia, were purchased from American Tissue Culture Collection.
Isolation and culture of peritoneal macrophages
Murine peritoneal macrophages were isolated by washing the peritoneal cavity with 10 ml of ice-cold phosphate buffered saline (PBS) supplemented with 1% fetal bovine serum. Individual cell suspensions were washed 2× with 1% fetal bovine serum–supplemented PBS. Murine peritoneal macrophages were plated at 1 × 106/ml in a 6-well plate and cultured by 1% fetal bovine serum–supplemented RPMI 1640 medium. Cells were left unstimulated as a control or stimulated with 10 ng/ml or 1000 ng/ml of lipopolysaccharide (LPS; Sigma-Aldrich) for 24 h at 37°C and 5% CO2.
Gene silencing of Fli1 in dermal fibroblasts and endothelial cells and quantitative reverse transcription-PCR (qRT-PCR)
For small interfering RNA (siRNA) experiments, cells were seeded shortly before transfection. The cells were transfected with 10 nM of FLI1 siRNA (Santa Cruz Biotechnology) or scrambled non-silencing RNA (Santa Cruz Biotechnology) using HiPerfect transfection reagent (Qiagen) for 72 h. Cells were then serum-starved for the last 24 h. mRNA levels of the FLI1 and CXCL6 genes in human cells and those of the Fli1 and CXCL6 genes in murine macrophages were examined by qRT-PCR and normalized to mRNA levels of the GAPDH or Gapdh genes. The sequences of primers are as follows: CXCL6-forward 5′-AGA GCT GCG TTG CAC TTG TT-3′, CXCL6-reverse 5′-GCA GTT TAC CAA TCG TTT TGG GG-3′; FLI1-forward 5′-GGA TGG CAA GGA ACT GTG TAA-3′, FLI1-reverse 5′-GGT TGT ATA GGC CAG CAG-3′; GAPDH-forward 5′-ACC CAC TCC TCC ACC TTT GA-3′, GAPDH-reverse 5′-CAT ACC AGG AAA TGA GCT TGA CAA-3′; Cxcl6-forward 5′-GCT GCC CCT TCC TCA GTC AT-3′, Cxcl6-reverse 5′-CAC CGT AGG GCA CTG TGG A-3′; Fli1-forward 5′-ACT TGG CCA AAT GGA CGG GAC TAT-3′, Fli1-reverse 5′-CCC GTA GTC AGG ACT CCC G-3′; Gapdh-forward 5′-CGT GTT CCT ACC CCC AAT GT-3′, Gapdh-reverse 5′-TGT CAT CAT ACT TGG CAG GTT TCT-3′.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the EpiQuik ChIP kit (Epigentek). After reversal of crosslinking, the immunoprecipitated chromatin was amplified by PCR of specific regions of target genes. The amplified DNA products were resolved by agarose gel electrophoresis. Putative Fli1 binding site was predicted by Tfsitescan. Primer sequences for the CXCL6 gene were as follows: CXCL6/F-346 5′-GAG GAG CAT CTC CCA GAC AG-3′, and CXCL6/R-193 5′-AAG GGG GAG GAC ATT TTA GC-3′. No nonspecific amplification was detected in these experiments.
Patients
Serum samples, frozen at −80°C until assayed, were obtained from 57 patients with SSc [53 women, 4 men; median age 59 yrs, interquartile range (IQR) 52–69.5; disease duration 5 yrs, 1.9–12] after getting informed consent. Patients treated with corticosteroids or other immunosuppressants prior to their first visits were excluded. Patients were grouped by the LeRoy’s classification system: 27 patients with dcSSc (age 58 yrs, IQR 48–63; disease duration 3 yrs, 1.5–10) and 30 patients with lcSSc (age 62 yrs, IQR 54–74; disease duration 9 yrs, 2–20). All patients fulfilled the new classification criteria of SSc31. For mRNA expression analyses of the whole skin, cDNA was prepared from the forearm skin samples of 27 patients with SSc, which consisted of a totally different patient population from those enrolled in the analysis of sera.
The measurement of serum CXCL6 levels
Specific ELISA kits (R&D Systems) were used to measure serum CXCL6 levels. Briefly, polystyrene 96-well plates coated with anti-CXCL6 antibodies were incubated with 50 μl of serum at room temperature for 2 h. Then, the wells were washed and incubated at room temperature for 2 h with horseradish peroxidase-conjugated anti-CXCL6 antibodies. Next, the wells were washed again, supplemented with tetramethylbenzidine, and incubated at room temperature for 30 min. Finally, sulfuric acid was added to terminate the reaction and the absorbance at 450 nm was measured. Serum CXCL6 levels were calculated using standard curve.
Clinical assessments
Disease onset was defined as the first clinical event that was a clear manifestation of SSc other than Raynaud phenomenon. The duration of disease was defined as the interval between the onset and the time of blood sampling. Skin score was measured using the modified Rodnan total skin thickness score (mRSS). The degree of interstitial lung disease (ILD) was evaluated by the percentage of predicted vital capacity (%VC) and the percentage of predicted DLCO (%DLCO) on pulmonary function test. Ground-glass opacity (GGO) score and fibrosis score of ILD were determined as previously described32. Digital ulcers (DU) and digital pitting scars (DPS) are defined as previously reported33. The details of assessment for organ involvement are described in the legend of Table 1.
Clinical correlation of serum CXCL6 levels in patients with SSc. Statistical analysis was carried out with a Fisher’s exact probability test for the analysis of frequency. Values are median (interquartile range), n unless otherwise specified.
Statistical analysis
Statistical analysis was carried out with the Mann-Whitney U test to compare the distributions of 2 unmatched groups, the 2-tailed paired Student t test for the comparison of normally distributed paired data, and a Fisher’s exact probability test for the analysis of frequency. Correlations with clinical data were assessed by Spearman rank correlation coefficient. The normal distribution was evaluated by the Shapiro-Wilk test. Multivariate regression analysis was conducted with stepwise procedure. Statistical significance was defined as a p value of < 0.05.
RESULTS
CXCL6 expression is directly regulated by transcription factor Fli1 in dermal fibroblasts, dermal microvascular endothelial cells, and macrophages
Previous reports demonstrated that CXCL6 is expressed by dermal fibroblast and endothelial cells stimulated with proinflammatory cytokines, such as IL-1β, and macrophages stimulated with LPS, but not peripheral blood mononuclear cells stimulated with LPS34. Because Fli1 deficiency induces SSc-like phenotypes in dermal fibroblasts, dermal microvascular endothelial cells, and macrophages in vivo and in vitro13,35, we initially looked at the effect of Fli1 deficiency on the expression levels of CXCL6 in those cells. When human dermal fibroblasts and HDMEC were treated with FLI1 siRNA or scrambled non-silencing RNA, CXCL6 mRNA expression was significantly increased in FLI1 siRNA-treated cells compared with scrambled non-silencing RNA-treated cells (Figure 1A and Figure 1B). On the other hand, when murine peritoneal macrophages isolated from Fli1+/− mice and control litter-mates were compared, LPS-dependent induction of Cxcl6 mRNA expression was significantly inhibited by Fli1 haploinsufficiency (Figure 1C). Importantly, Fli1 occupied the CXCL6 promoter in human dermal fibroblasts, HDMEC, and THP-1 cells (Figure 1D). To further confirm whether Fli1 deficiency truly recapitulates CXCL6 expression in SSc, we looked at CXCL6 mRNA expression in SSc and normal dermal fibroblasts. As shown in Figure 1E, SSc dermal fibroblasts exhibited a significantly higher expression of CXCL6 mRNA than normal dermal fibroblasts. On the other hand, the stimulation of TGF-β, a profibrotic growth factor involved in the activation of SSc dermal fibroblasts, unexpectedly suppressed CXCL6 mRNA expression in normal dermal fibroblasts (0.15 ± 0.02× relative to the baseline, p = 0.004). Taken together with the evidence that CXCL6 expression is increased in SSc endothelial cells29, these results indicate that CXCL6 is a target molecule of transcription factor Fli1 in fibroblasts, endothelial cells, and macrophages, and that Fli1 deficiency contributes to CXCL6 upregulation in SSc dermal fibroblasts and SSc dermal microvascular endothelial cells.

Fli1 deficiency regulates CXCL6 expression in fibroblasts, endothelial cells, and macrophages. (A, B) The expression of CXCL6 and FLI1 mRNA was evaluated by qRT-PCR in (A) human dermal fibroblasts (n = 8) and (B) HDMEC (n = 6) treated with FLI1 small interfering RNA or SCR. (C) Cxcl6 mRNA levels were determined by qRT-PCR in murine Fli1+/− and WT peritoneal macrophages treated with LPS (n = 3). (D) CXCL6 mRNA expression was assessed by qRT-PCR in SSc and normal dermal fibroblasts (n = 5). (E) The binding of Fli1 to the CXCL6 promoter was examined by chromatin immunoprecipitation in human dermal fibroblasts, HDMEC, and THP-1 cells. For qRT-PCR, results of controls or relative value compared with the controls are expressed as mean ± SD. Statistical analysis was carried out with the 2-tailed paired Student t test for (A) to (C) and with the Mann-Whitney U test for (D). For chromatin immunoprecipitation, representative results of 3 independent experiments are shown. * p < 0.05. Fli1: Friend leukemia virus integration 1; qRT-PCR: quantitative reverse transcription-PCR; HDMEC: human dermal microvascular endothelial cells; SCR: scrambled non-silencing RNA; siRNA: small interfering RNA; WT: wild-type; LPS: lipopolysaccharide; SSc: systemic sclerosis; AU: arbitrary unit; IgG: immunoglobulin G; HC: healthy controls.
Serum CXCL6 levels positively correlate with clinical features related to tissue fibrosis in patients with SSc
To further investigate whether altered CXCL6 expression contributes to the development of SSc, we evaluated the clinical correlation of CXCL6 levels in the skin and sera because a previous report demonstrated that CXCL6 expression in endothelial cells and sera are elevated in patients with SSc compared with healthy controls29. We first focused on the fibrotic aspect of SSc. When looking at mRSS, a significant positive correlation with serum CXCL6 levels was detected in total patients with SSc (r = 0.31, p = 0.022; Figure 2A). In addition, CXCL6 mRNA levels in the lesional skin significantly correlated with mRSS in patients with SSc (r = 0.55, p = 0.003; Figure 2B). With respect to ILD, its presence was associated with a significant elevation of serum CXCL6 levels (Table 1). Importantly, serum CXCL6 levels inversely correlated with %VC and %DLCO in total patients with SSc (r = −0.36, p = 0.0067 for %VC, Figure 2C; r = −0.29, p = 0.037 for %DLCO, Figure 2D). Further, in patients with SSc with ILD, serum CXCL6 levels showed an inverse correlation with the fibrosis score (r = 0.45, p = 0.0091; Figure 2E), while not with GGO score (r = 0.14, p = 0.45), suggesting that CXCL6 is associated with the resultant fibrosis, but not currently progressive alveolitis of SSc-associated ILD. As for heart involvement and esophageal dysfunction, both of which are related to tissue fibrosis, the presence of heart involvement, but not esophageal dysfunction, was linked to the elevation of serum CXCL6 levels (Table 1). These results suggest that CXCL6 is involved in the fibrotic process of the skin, lung, and heart in SSc.
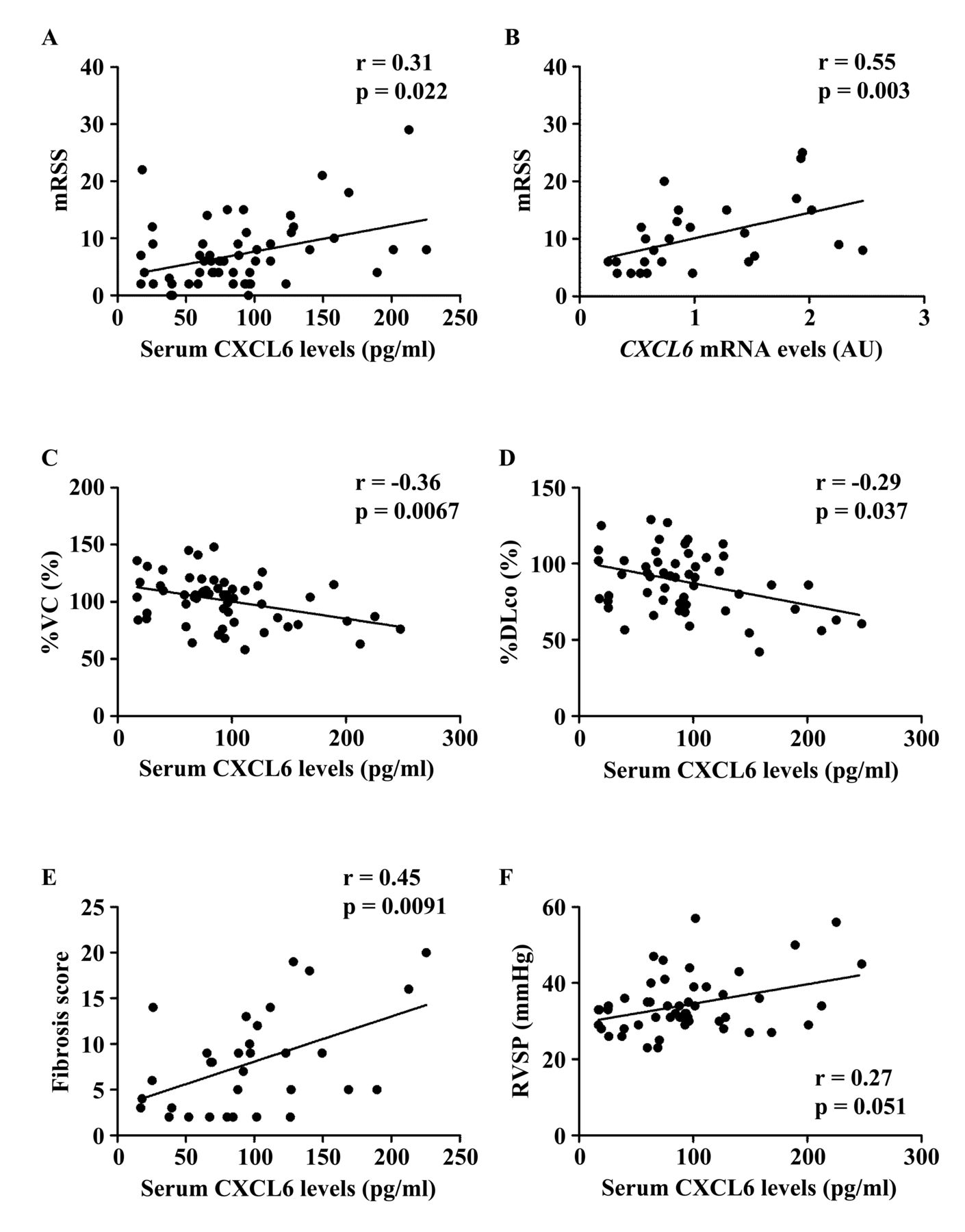
Correlation of CXCL6 levels in the skin and sera with clinical variables in patients with SSc. (A, C–E) Serum CXCL6 levels positively correlated with (A) the mRSS, (C) the %VC, (D) the %DLCO in total patients with SSc, and (E) with fibrosis score of chest computed tomography in SSc patients with interstitial lung disease. (B) There was a significant positive correlation between CXCL6 mRNA expression in the lesional skin and mRSS. (F) Serum CXCL6 levels tended to correlate with the values of RVSP in total patients with SSc. Statistical analysis was carried out with Spearman rank correlation test. SSc: systemic sclerosis; mRSS: modified Rodnan skin score; %VC: percentage of predicted vital capacity; %DLCO: percentage of the predicted DLCO; RVSP: right ventricular systolic pressure.
Serum CXCL6 levels are elevated in patients with SSc with vascular complications
We further examined whether vascular complications affect serum CXCL6 levels in patients with SSc. We focused on cutaneous vascular manifestations, such as Raynaud phenomenon, nailfold bleeding, telangiectasia, and DU/DPS. Of note, patients with these symptoms except nailfold bleeding had significantly elevated serum CXCL6 levels compared with corresponding patients without each symptom (Table 1). Regarding organ involvement related to proliferative obliterative vasculopathy, we assessed the association of serum CXCL6 levels with elevated right ventricular systolic pressure (RVSP) and scleroderma renal crisis. Serum CXCL6 levels were significantly greater in patients with elevated RVSP than in those without (Table 1). Of note, there was a trend toward a positive correlation between serum CXCL6 levels and RVSP values (r = 0.27, p = 0.051; Figure 2F). Regarding scleroderma renal crisis, we could not draw a definitive conclusion because only 2 patients had a history of this complication. These results indicate that CXCL6 is likely to contribute to the development of a variety of cutaneous and visceral vascular complications associated with SSc.
Serum CXCL6 levels are predominantly related to the presence of DU/DPS and heart involvement in patients with SSc
As described above, serum CXCL6 levels correlated with a variety of clinical symptoms in patients with SSc. Although the correlation of serum CXCL6 levels with numeric variables, such as mRSS, pulmonary function test results, and scores of GGO and fibrosis, was strictly assessed by Spearman rank correlation coefficient, it remained unclear in the case of qualitative variables (presence or absence of symptoms) which symptoms truly associate with serum CXCL6 levels in SSc. To address this issue, we conducted multivariate regression analysis with stepwise procedure using serum CXCL6 levels as the dependent variable and clinical symptoms with p values of < 0.05 in Table 1 as independent variables. To convert qualitative data to quantitative values, we used dummy variables, namely, patients were given a value of 1 if they had the symptom or a value of 0 if they did not have the symptom. As shown in Table 2, the final model included DU/DPS and heart involvement as independent variables. These results indicate that CXCL6 may be mainly included in the developmental process of DU/DPS and heart involvement in patients with SSc.
Factors predicting serum CXCL6 levels determined by multivariate regression analysis.
DISCUSSION
In our present study, we demonstrated the enhanced expression of CXCL6 in dermal fibroblasts and endothelial cells and the suppressed expression of CXCL6 in macrophages under a Fli1-deficient condition. Given that Fli1 deficiency induces SSc-like phenotypes in those cells, these results suggest that CXCL6 is involved in the development of fibroblast- and endothelial cell–mediated clinical symptoms of SSc. Supporting this notion, further analyses with sera of patients with SSc revealed the association of circulating CXCL6 levels with a variety of fibrotic and vascular clinical symptoms. Taken together with the evidence that neutrophil infiltration is rarely seen in SSc lesional skin despite increased expression of CXCL6 in dermal microvascular endothelial cells29, these results suggest that CXCL6 serves as a potent proangiogenic factor rather than a neutrophil chemoattractant in SSc, possibly leading to aberrant vascular activation and subsequent tissue fibrosis. Overall, the present data reinforce the previous finding regarding the contribution of CXCL6 to the vascular aspect of SSc pathogenesis29.
A possible involvement of CXCL6 in the pathological tissue fibrosis has been reported in human diseases and animal models. For instance, CXCL6 concentration is increased in bronchoalveolar lavage fluid from patients with idiopathic pulmonary fibrosis and in the lung of mice exposed to bleomycin. Of note, anti-CXCL6 antibody suppresses acute inflammation and late-phase pulmonary fibrosis in bleomycin-treated mice36. This seems plausible because neutrophils are abundantly seen in lung tissues of these pathological conditions. On the other hand, in human chronic hepatitis C virus infection CXCL6 is nominated as a member of genes upregulated along with the progression of stage and collagen deposition37,38,39. Given that lymphocytes, macrophages, and endothelial cells, but not neutrophils, are implicated in fibrosis progression of liver40, endothelial cell activation seems to be involved with CXCL6 in this pathological condition. Despite these accumulating data, however, the detailed mechanism by which CXCL6 regulates the pathological process underlying tissue fibrosis is still unclear. In our present study, serum CXCL6 levels correlated inversely with %VC and %DLCO and positively with mRSS and the fibrosis score. In addition, CXCL6 mRNA levels in the lesional skin revealed a positive correlation with mRSS. These current data suggest that CXCL6 is involved in the development of dermal and pulmonary fibrosis in SSc. Importantly, serum CXCL6 levels did not correlate with GGO score, therefore it is likely that CXCL6 is involved preferentially in the induction of tissue fibrosis rather than inflammation. Because CXCR2, a receptor for CXCL6, is expressed in only ∼2% of α-smooth muscle actin–positive SSc fibroblasts, CXCL6 seems not to be a major driver of fibroblast activation41. Taking into account the close association of vasculopathy with tissue fibrosis in SSc42, these findings further strengthen the hypothesis that the effect of CXCL6 on endothelial cells contributes to the development of dermal and pulmonary fibrosis in SSc.
Through multivariate regression analysis with stepwise procedure, we identified DU/DPS and heart involvement as independent factors predicting serum CXCL6 levels. According to a recent study regarding SSc heart involvement, cardiac fibrosis is paralleled by microvascular endothelial cell apoptosis and reduced capillary density43. Also, DU/DPS belong to a set of cutaneous symptoms closely associated with loss of capillaries because the decreased capillary density in nailfold videocapillaroscopic evaluation is a risk factor for the development of new DU44. Therefore, these results suggest that CXCL6 expression is related to the induction of endothelial cell apoptosis. Importantly, SSc endothelial cells show aberrant response to CXCL6 stimulation, such as increased phosphorylation of c-Jun N-terminal kinase and p38, death signaling pathways, and decreased phosphorylation of AKT, a cell survival signaling pathway, suggesting that CXCL6 promotes the apoptosis of SSc endothelial cells29. Therefore, it is plausible that DU/DPS and heart involvement were identified as independent predictive factors for elevated serum CXCL6 levels in SSc.
The association of endothelial cell apoptosis with tissue fibrosis has been reported in SSc. First, loss of capillaries because of endothelial cell apoptosis generally induces tissue hypoxia, leading to the activation of fibroblasts in SSc45. Second, apoptotic endothelial cells release soluble factors, such as a C-terminal fragment of the domain V of perlecan, promoting cell survival and myofibroblastic differentiation of interstitial fibroblasts46. Third, it is speculated that self-antigens released from apoptotic endothelial cells bind to LL-37, and the complex subsequently promotes the production of IFN-γ from plasmacytoid dendritic cells through Toll-like receptor 7 and 9. This speculation is proposed based on the following findings: (1) chemerin, a potent chemoattractant for plasmacytoid dendritic cells, and LL-37 are abundantly expressed in SSc dermal small vessels17,21, (2) plasmacytoid dendritic cells are distributed predominantly in the perivascular regions and highly express IFN-γ in SSc lesional skin47, and (3) IFN-γ induces vascular injury and senescence as well as autoimmune reaction, leading to fibroblast activation48. Importantly, Fli1 deficiency induces the expression of chemerin and LL-37 in endothelial cells in vivo and in vitro17,21. Therefore, Fli1 deficiency–dependent induction of endothelial CXCL6 expression, which potentially induces endothelial cell apoptosis, further supports the idea that Fli1 deficiency is a critical factor integrating various pathological events involved in the development of SSc vasculopathy10,35,49.
We documented a series of data confirming a possible contribution of CXCL6 to the development of SSc. Consistent with a previous study, CXCL6 is likely to be involved in endothelial cell apoptosis, leading to the development of vasculopathy and tissue fibrosis in SSc. The association of Fli1 deficiency with CXCL6 upregulation in endothelial cells further supports the notion that Fli1 deficiency is a critical predisposing factor of SSc.
Acknowledgment
We thank T. Kaga, A. Hatsuta, C. Ohwashi, T. Ikegami, and N. Watanabe for technical assistance.
Footnotes
Full Release Article. For details see Reprints and Permissions at jrheum.org
Supported by a grant for Research on Intractable Diseases from the Ministry of Health, Labor, and Welfare of Japan.
- Accepted for publication April 17, 2017.








{kind=link}
{kind=link}