

Abstract
Objective. Inflammatory diseases, specifically rheumatoid arthritis (RA), are assumed to increase the risk of coronary artery disease (CAD). More recently, multiple single-nucleotide polymorphisms (SNP) associated with RA risk were identified. If causal mechanisms affecting risks of RA and CAD are overlapping, risk alleles for RA might also increase the risk of CAD.
Methods. Sixty-one SNP associating with RA in genome-wide significant analyses were tested for association with CAD in CARDIoGRAM (Coronary ARtery DIsease Genome wide Replication and Meta-analysis), a metaanalysis including genome-wide association data (22,233 CAD cases, 64,762 controls). In parallel, a set of SNP being associated with low-density lipoprotein cholesterol (LDL-C) was tested as a positive control.
Results. Twenty-nine RA-associated SNP displayed a directionality-consistent association with CAD (OR range 1.002–1.073), whereas 32 RA-associated SNP were not associated with CAD (OR range 0.96–0.99 per RA risk-increasing allele). The proportion (48%) of directionality-consistent associated SNP equaled the proportion expected by chance (50%, p = 0.09). Of only 5 RA-associated SNP showing p values for CAD < 0.05, 4 loci (C5orf30, IL-6R, PTPN22, and RAD51B) showed directionality-consistent effects on CAD, and 1 (rs10774624, locus SH2B3) reached study-wide significance (p = 7.29E-06). By contrast, and as a proof of concept, 46 (74%) out of 62 LDL-C–associated SNP displayed a directionality-consistent association with CAD, a proportion that was significantly different from 50% (p = 5.9E-05).
Conclusion. We found no evidence that RA-associated SNP as a group are associated with CAD. Even though we were not able to study potential effects of all genetic variants individually, shared nongenetic factors may more plausibly explain the observed coincidence of the 2 conditions.
Inflammation is involved in the pathophysiology of coronary artery disease (CAD)1. Interestingly, primarily inflammatory diseases such as rheumatoid arthritis (RA) or systemic lupus erythematosus have been found to be associated with a markedly enhanced risk of CAD2,3. For example, in 1 study the risk of being affected by CAD was 3-fold higher in women with RA compared with controls4. Moreover, patients with RA showed a prevalence of subclinical femoral atheromatosis comparable to patients with diabetes5, indicating that RA may be an additional independent factor for the development of atherosclerotic diseases. Further, epidemiological studies have reported that RA increases cardiovascular (CV) mortality6,7. In autopsy studies, patients with RA showed more vulnerable plaques and more inflammation in the coronary artery walls, but a less severe plaque burden compared with controls8. Coronary spasm and changes of endothelial function were subsequently discussed to affect cardiac outcome rather than coronary obstruction9.
More recently, substantial information has been generated on genetic factors contributing to the complex nature of RA. Based on genome-wide association data, multiple single-nucleotide polymorphisms (SNP) have been reported to be associated with RA at a genome-wide significance level10,11,12,13,14,15,16,17. Despite the epidemiological and pathophysiological evidence for an association of RA with CAD, there is still uncertainty whether genetically mediated mechanisms that increased the risk for RA also affect the risk of CAD3.
In our present analysis, we assessed whether RA and CAD have overlapping genetic roots by testing whether RA-associated SNP are also associated with an increased risk of CAD. We performed our analyses in the CARDIoGRAM (Coronary ARtery DIsease Genome wide Replication and Meta-analysis) dataset, which includes genome-wide association data from 22,233 patients with CAD and 64,762 population-based controls18.
MATERIALS AND METHODS
Study sample
The CARDIoGRAM consortium combined genome-wide association data on CAD cases and controls from 14 patient-based and population-based studies and consortia in a large metaanalysis: Cohorts for Heart and Aging Research in Genomic Epidemiology19, CADomics, Atherosclerotic Disease VAscular functioN and genetiC Epidemiology study20, deCODE CAD study21, Ludwigshafen Risk and Cardiovascular Health Study22/AtheroRemo 1 and 2, Myocardial Infarction Genetics Consortium23, MedStar24, Ottawa Heart Genomics Study25, PennCATH24, the Wellcome Trust Case Control Consortium (WTCCC)26, and the German Myocardial Infarction Family Studies (GerMIFS) I, II, and III26,27,28. The protocol of each participating study was approved by their local ethics committees. In addition, our current analyses were approved by the ethics committee of the Technical University of Munich (ID 29516). The main findings of our metaanalysis have been reported elsewhere29. In brief, using data from 22,233 CAD cases and 64,762 controls, our analysis identified 25 loci to be genome-wide significantly associated with CAD. Risk alleles at these 25 loci increased CAD risk by 6% to 17%. A detailed description of the main results and individuals (cases/controls) in the participating studies is presented in the supplementary data (Supplementary Table 1 and Supplementary Table 2, available from the authors on request).
The statistical analysis plan for the metaanalyses of the CARDIoGRAM consortium has been described elsewhere in detail18. In brief, the associations between SNP and CAD were tested within each study group using a logistic regression model adjusting for age, sex, and potential population stratification, assuming an additive genetic model, which is often used in genome-wide association study (GWAS) settings analyzing common diseases. Then a metaanalysis was performed based on the study-specific effect estimates and their standard errors, and using random-effects or fixed-effects models depending on heterogeneity between the studies. Observed associations between RA SNP and CAD are reported as OR and their 95% CI.
SNP selection
Genetic variants associated with RA in whites were identified using the GWAS catalogue from the National Human Genome Research Institute (www.genome.gov/gwastudies) and were also reported by Eyre, et al16.
In the current literature, we identified 65 SNP displaying association with RA on a genome-wide significant level10,11,12,13,14,15,16,17. For SNP that were not available or did not pass quality control in CARDIoGRAM, we identified proxy SNP. We searched for proxies with an R2-threshold of 0.8 or higher and a maximal distance of 500 kb. The identified proxy SNP were then tested for association with CAD in CARDIoGRAM. Four SNP of the initially identified variants were not represented in CARDIoGRAM and we could not find appropriate proxies. In total, we integrated 61 genetic variants in our analysis. In secondary analyses, we excluded 38 RA-associated SNP with potential pleiotropic effects on other CAD risk factors (e.g., diabetes, triglycerides, blood pressure, high-density lipoprotein, other inflammatory conditions; Supplementary Table 3, available from the authors on request) and repeated the association analyses with CAD with the remaining RA-associated SNP. Finally, we restricted our analysis to a CARDIoGRAM subgroup containing only women (6579 cases and 23,770 controls) because women are more often affected by RA30. By doing so, we intended to see whether the association between genetic variants and CAD was different between men and women (effect modification by sex).
For our proof-of-concept analysis, we tested 62 SNP known to associate with low-density lipoprotein cholesterol (LDL-C) at a genome-wide significance level31,32 for their association with CAD.
Moreover, we searched the literature for non-HLA SNP, which have been associated with RA disease severity in recent reports33,34,35, and subsequently tested these for its association with CAD.
Statistical methods
First, we assessed the proportion of RA-associated SNP that displayed a directionality-consistent association with CAD (CAD OR > 1 per RA risk-increasing allele, irrespective of statistical significance of the individual SNP). If RA represented a causal risk factor for CAD, we expected the proportion of RA risk-increasing alleles, which were also positively associated with CAD, to be significantly greater than 50%. This hypothesis was assessed using an exact binomial test. Second, we compared the observed effects of RA-associated SNP on CAD (observed in CARDIoGRAM) to the expected effects of these SNP on CAD. The published OR for CAD in patients with RA ranged from 1.6 to 2.036,37,38, with the largest population-based sample revealing an OR of 1.6. Given such an OR for the association of RA to CAD on the one hand and the respective effect of each SNP on RA on the other hand, we calculated the expected effects of RA SNP on CAD.
Risk score calculation
In addition to this approach, we tested whether the risk allele distribution of 61 RA-associated SNP differed between CAD cases and controls. To this end, we used individual level genotypes of 7 European studies (GerMIFS I–V, WTCCC-CAD, and Cardiogenics)26,27,28,29 with a total of 8542 CAD cases and 10,273 controls and calculated a genetic risk score as follows: for each individual we modeled the 61 SNP as a multilocus genetic risk score by summing the number of risk alleles (0/1/2) for each of the SNP, weighted by their estimated RA effect sizes as reported in the literature. We performed a simple Student t test to compare the differences of the weighted risk score between CAD cases and controls in each study, and then calculated a combined p value by a weighted Z score based on sample size.
RESULTS
Association of RA-associated SNP with CAD
Of the 61 analyzed RA SNP, about half (n = 29) showed an OR > 1 for prevalent CAD in CARDIoGRAM (OR range 1.002–1.073), whereas the other RA-associated SNP (n = 32) displayed an OR < 1 (OR range 0.96–0.99; Figure 1). Thus, this distribution was not different from that expected by chance (50%, p = 0.09).
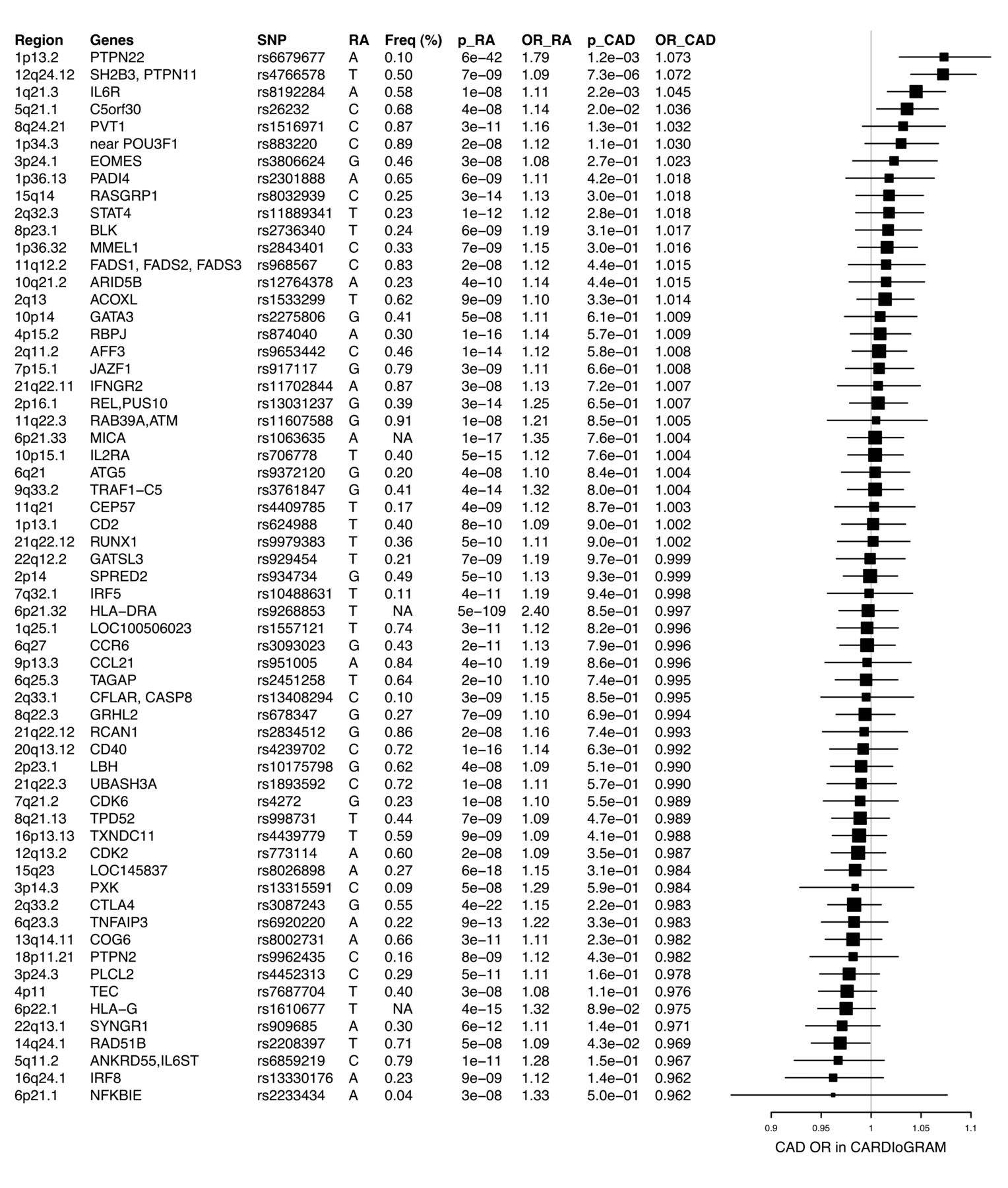
Genes and respective lead SNP (rs numbers) are listed. Forest plots display the associations of RA associated SNP with CAD in CARDIoGRAM. Boxes represent the OR and whiskers 95% CI. SNP: single-nucleotide polymorphism; RA: rheumatoid arthritis; CAD: coronary artery disease.
Four SNP within the loci of C5orf30, SH2B3, IL-6R, and PTPN22 showed directionality-consistent effects on CAD and had p values < 0.05. The SNP (rs10774624, p = 7.29E-06) at the SH2B3 locus also reached study-wide significance (p = 0.05 ÷ 61 = 0.0008). One SNP (rs2208397) within RAD51B had a p value < 0.05 (without Bonferroni correction), but the risk alleles were not the same for RA and CAD.
Secondary analyses
After removing all SNP with potential effects on traditional CAD risk factors (n = 37), the results did not change remarkably. Eleven out of 24 SNP (46%) produced OR greater than 1, while the remaining 13 SNP had CAD OR below 1 (54%, p = 0.15). Only 1 SNP (rs26232 at the C5orf30 locus) remained, showing directionality-consistent effects on CAD with a nominally (not study-wide) significant p value (p = 0.02; Supplementary Figure 1, available from the authors on request).
Because RA is more prevalent among women30, we performed an analysis only in women in CARDIoGRAM (6579 cases and 23,770 controls). Out of 57 SNP available, 24/57 SNP (42%) displayed directionality-consistent effects on CAD and RA (p = 0.052). None of the SNP produced a p value < 0.05 (Supplementary Figure 2, available from the authors on request).
Comparison of expected and observed effects of RA-associated SNP on CAD
We calculated the expected effects of RA-related SNP on CAD risk using an assumed effect size (β) of 0.47 of RA on CAD38. The expected OR ranged from 1.04 to 1.51 (Figure 2). We found that the observed effects on CAD (range 0.962–1.073) differed statistically significantly (p = 4.9e-13) from the expected effects.

Bar plot comparing the predicted and observed effects on CAD. The predicted effects are calculated based on the respective SNP effects on RA and the effect of RA on CAD coming from epidemiological data. The observed effects were derived from the CARDIoGRAM dataset. CAD: coronary artery disease; SNP: single-nucleotide polymorphism; RA: rheumatoid arthritis.
Association of LDL-C–associated SNP with CAD
As a positive control for our methodological approach, we assessed the association of LDL-C–associated genetic variants (thus, variants that were associated with an established causal risk factor for CAD) for association with CAD31,32. As expected, most LDL-C–increasing alleles (n = 46 of n = 62, 74%) displayed a positive association with CAD (a proportion that differed statistically significantly from the 50% expected by chance, p = 5.9E-05; Supplementary Figure 3, available from the authors on request).
Weighted genetic risk score
Using individual-level data from a total of 8542 CAD cases and 10,273 controls, we assessed whether the distribution of RA-associated risk alleles differed between CAD cases and controls. The mean genetic RA score in CAD cases (8.37) was not statistically different from the mean score in controls (8.35, p = 0.26).
SNP associated with RA disease severity
In total, we identified 19 SNP in more recent reports, which have been associated with radiographic disease progression. For 7 SNP, conflicting results regarding the effect allele exist. Hence, we tested a set of 12 SNP, of which 9 SNP produced OR > 1 (p = 0.05 using an exact binomial test). One of these SNP (rs1528873) at the DKK-1 locus was associated nominally significantly with CAD risk with directionality-consistent effects (Supplementary Table 4, available from the authors on request).
DISCUSSION
Using data from 22,233 patients with CAD and 64,762 controls, we assessed the association of RA-related SNP with CAD. We found no evidence for a statistically significant association of RA SNP with CAD risk. About half of the RA-associated SNP (n = 29, 48%) displayed CAD OR greater than 1, while the other half (n = 32 SNP) produced OR below 1 for the association with CAD in our dataset, a distribution as expected by chance. There was, however, one remarkable exception. An SNP (rs10774624) at the SH2B3 locus showed directionality-consistent effects on CAD and reached study-wide significance (p = 7.29E-06). Indeed, associations between the SH2B3 locus and CAD have been reported previously29. However, this effect is possibly driven by pleiotropic effects on, for example, blood pressure or Type 1 diabetes39,40. In line with this notion, the encoded protein of this locus (SH2B adaptor protein 3) has been found to enhance inflammation and affect the vascular biology and homeostasis by complex intracellular signaling pathways41.
After removing all SNP with potential effects on CV risk factors, the results did not change substantially. Similarly, restricting our analysis to women did not affect the results. In addition, the burden of RA risk alleles (expressed as a weighted genetic risk score or RA) was not different between CAD cases and controls. As a proof of principle, we analyzed 62 genetic variants associated with LDL-C — a causal risk factor for CAD — in the same fashion. As expected, most SNP (46/62, 74%) displayed a positive association with CAD.
The clinical association between RA and CAD has been recognized in many prior studies4. Compared with healthy individuals, patients with RA have an increased risk of dying from CV causes42, have an increased risk of clinical CVD events (such as CAD42 and stroke43), and present more often with subclinical CV disease (CVD) manifestations, as indicated, e.g., by an altered pulse wave velocity, increased carotid intima-media thickness (IMT), and lower flow-mediated dilatation as compared with controls (reviewed44). Guidelines, therefore, suggest to screen patients with RA for CAD and subclinical vascular pathologies and to treat them early and aggressively once atherosclerosis is detected45.
However, whether RA actually causes CAD has not been proven. Prior genetic analyses evaluated defined genetic variation as independent risk factors for CV events in patients with RA. For example, CD40, which acts as a key player of the inflammation process initiation, carries SNP at its locus that have been found to associate with RA itself and with carotid IMT, a marker for subclinical CVD, in patients with RA46. Further, genetic variants of the interferon regulatory factor 5 were found to be associated with an increased risk of CV events in patients with RA47.
In our larger dataset, we found no supporting evidence for an association of genetic variants at the above-mentioned individual loci with CAD risk.
We expanded on these analyses by using large genome-wide datasets to assess whether risk alleles that are associated with an increased risk for RA are also associated with CAD risk. The underlying concept is that if RA is causally related to CAD, RA-related alleles would also translate into observable increases in CAD risk. However, in our large dataset including 22,233 CAD cases and 64,762 controls, we found no such evidence.
As an alternative explanation for the greater CV risk in patients with RA, a greater burden with traditional risk factors among patients with RA48 has been emphasized by many authors. Indeed, arterial hypertension and unfavorable lipid profiles have been found to be more prevalent in patients with RA than in controls49. Other reports provided some evidence that the increased CVD risk could be because of side effects of RA medication. In particular, the common use of nonsteroidal antiinflammatory drugs, cyclooxygenase-2 inhibitors, and oral glucocorticoids has been shown to significantly increase systolic blood pressure, which might subsequently result in a higher risk of CVD50. Despite the evidence for an increased prevalence of traditional risk factors in patients with RA, some authors argue that traditional factors even in accumulation are not able to fully explain the increased risk of CAD46. In this regard, an underlying inflammatory background linking RA and CAD has been frequently pronounced. This concept is supported by growing evidence of reduced CV risk, associated with using antiinflammatory agents such as methotrexate or biologicals such as antitumor necrosis factor-α46.
Another observation deserves some consideration. The incidence of CAD in patients with RA may be influenced by the presence of anticitrullinated peptide antibodies (ACPA): studies reported a higher risk of CAD in ACPA-positive patients compared with ACPA-negative patients with RA51. Interestingly, the genetic background of these 2 RA subtypes shows a remarkable difference. While the specific HLA-DRB1 alleles are commonly found in ACPA-positive patients, ACPA-negative patients often lack this important genetic risk factor52,53. Overall, however, ACPA-negative and ACPA-positive RA share a large proportion of the reported susceptibility loci54. Hence, we do not feel that this specific difference has influenced our principal findings.
SNP associating with RA susceptibility produced a null finding in our overall dataset. Interestingly, previous reports have shown that many SNP that are associated with the risk of contracting RA are not necessarily associated with disease severity35.
We therefore specifically studied SNP known to associate with disease severity and observed a trend for a positive association with CAD risk in CARDIoGRAM (9 out 12 SNP produced OR > 1). Only susceptibility variants in the HLA region were associated with both disease susceptibility and severity in several analyses33. By contrast, non-HLA variants did not produce such significant results in radiographic disease progression33. Our data indeed indicate that SNP that are associated with the risk of developing RA are not associated with CAD. However, SNP that are associated with greater inflammatory activity in patients with RA (more severe disease activity) provided some evidence of association with CAD in CARDIoGRAM, underscoring the concept that it is rather the inflammatory burden associated with (increased) RA disease activity that predisposes to CAD than the RA disease per se.
Strengths and limitations
The large and genome-wide dataset of CARDIoGRAM allowed us to assess many (n = 61) SNP for their association with CAD that have previously been identified to associate with RA at a genome-wide significant level. The following limitations merit consideration. First, 4 RA SNP did not pass quality control in CARDIoGRAM and we did not find appropriate proxy SNP. Although very unlikely, data from these missing SNP could have theoretically altered our conclusions. Second, we were not able to study the relation of RA-associated SNP with clinical RA in our dataset. Third, based on a reported prevalence of 0.8% in the general adult population55, about 700 participating individuals in CARDIoGRAM might be affected by RA. Fourth, many studies contributing to CARDIoGRAM included younger patients with myocardial infarction and CAD only. Hence, the effect of RA on CAD might not have been identified adequately in these individuals because RA might act as a risk factor for CAD rather later in life. Fifth, it is possible that some RA-associated SNP exert unknown pleiotropic effects on intermediate CV phenotypes and thereby influence (neutralize) the risk of CAD. However, given the large number of SNP studied, it is unlikely that a systematic effect by these SNP on CAD might have altered our results significantly. Sixth, while designing the CARDIoGRAM database, many individual SNP of the HLA region were not considered. Not surprisingly, about two-thirds of individual HLA-DRB variants identified to associate with RA risk in GWAS (p < 5 × 10−08) were not represented in CARDIoGRAM. Therefore, we used rs9268839 as 1 significant representative in our analysis.
Taken together, our data provide no evidence for a causal association of RA with CAD risk. Together with a shared inflammatory background of CAD and RA, traditional risk factors might explain the increased prevalence of CAD in patients with RA more than a common genetic architecture.
APPENDIX 1.
List of study collaborators. Members of the CARDIoGRAM Consortium: Sekar Kathiresan, Muredach P. Reilly, Nilesh J. Samani, Heribert Schunkert, Jeanette Erdmann, Themistocles L. Assimes, Eric Boerwinkle, Alistair Hall, Christian Hengstenberg, Inke R. König, Reijo Laaksonen, Ruth McPherson, John R. Thompson, Unnur Thorsteinsdottir, Andreas Ziegler, Devin Absher, Li Chen, L. Adrienne Cupples, Eran Halperin, Mingyao Li, Kiran Musunuru, Michael Preuss, Arne Schillert, Gudmar Thorleifsson, Benjamin F. Voight, George A. Wells, Panos Deloukas, Hilma Holm, Robert Roberts, and Alexandre F.R. Stewart.
- Accepted for publication September 9, 2016.








{kind=link}
{kind=link}