

Abstract
Objective. While genetic risks have been implicated in systemic lupus erythematosus (SLE), the involvement of various genotypes in neuropsychiatric SLE (NPSLE) remains uncertain. The present metaanalysis aimed to combine data from different studies and evaluate the association between each genotype and the risk of developing NPSLE.
Methods. Studies were searched and retrieved from online databases (PubMed, EMBASE, BIOSIS, and ScienceDirect). Case-control studies were chosen if they reported genotype frequencies of the γ Fc region (FCγR) receptors II-A, III-A, and III-B; tumor necrosis factor–α (TNF-α); mannan-binding lectin (MBL); integrin alpha M (ITGAM); interleukin (IL) 1, IL-1β, and IL-6; IL-10 promoter; and vitamin D genes. The OR were used to assess the strength of this association between patients with NPSLE and SLE.
Results. A total of 33 studies were considered in this metaanalysis. The results suggest that these genotypes demonstrated a significant association with NPSLE: the homozygous FCγR IIIa 158 FF genotype (OR 1.89, p = 0.03 for FF vs VV + FV), heterozygous FCγR IIIb NA1/2 genotype (OR 2.14, p = 0.03 for NA1/2 vs NA1/1; OR 1.81, p = 0.04 for NA1/2 vs NA1/1 + NA2/2), and homozygous ITGAM rs1143679 HH genotype (OR 3.39, p = 0.04 for HH vs RH; OR 3.11, p = 0.048 for HH vs RR + RH). Polymorphisms of the TNF-α, MBL2, IL-1, IL-1β, IL-6, IL-10 promoter, and vitamin D receptor genes did not show a statistically significant association with the risk of developing NPSLE (p > 0.05).
Conclusion. This metaanalysis indicates that polymorphisms in the pathways of immune complex clearance, such as the FcγRIIIa, FcγRIIIb, and ITGAM genotypes, are potential susceptibility genes for NPSLE.
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease that is characterized by a higher prevalence in women than in men (female:male 9:1), loss of immunological tolerance to self-nuclear antigens, and abnormal B cell and T cell response1. Neuropsychiatric SLE (NPSLE) is one of the major and most damaging presentations2 of SLE. The American College of Rheumatology (ACR) research committee devised a nomenclature providing case definitions for 19 NP syndromes in SLE3. The ACR criteria include a wide range of neurological syndromes affecting the central (e.g., headache, meningitis, and seizure), peripheral (e.g., neuropathy), and autonomic nervous systems, as well as psychiatric syndromes (e.g., anxiety, cognitive dysfunction, and depression).
The heritability of SLE is suggested to be 66%, but current understandings of genetic variants explain 10–20% of SLE heritability4. The concordance rate of SLE in monozygotic twins (24–56%) is higher than the rate of dizygotic twins (2–4%)5. Similar to other psychiatric disorders such as schizophrenia, bipolar disorder, and Alzheimer’s disease, SLE is a disease of polygenic inheritance. The evidence of genetic susceptibility for SLE is mainly from genome-wide association studies, which have identified 40 disease-associated genes that are linked to SLE4. Mok and Lau6 estimated that at least 4 susceptibility genes are required for the development of SLE. These susceptibility genes are classified as HLA genes (e.g., DQA1 and DQB1) and non-HLA genes [e.g., γ Fc region (Fcγ) RIIA, interleukin (IL) 6, IL-10 promoter, mannose-binding lectin (MBL), and plasminogen activator inhibitor 1 (PAI-1)]7. Genetic polymorphisms in the IL-10 promoter, MBL, and PAI-1 genes may be associated with lupus nephritis in Chinese patients with SLE6,8,9,10. Chong, et al11 proposed that IL-10 causes the hyperactivity of B cells in SLE, and IL-10 is implicated in the pathogenesis of NPSLE12.
Accumulating studies suggest that susceptibility genes contribute to the NP manifestations in SLE. Koga, et al13 combined the risk alleles of 7 genes (BLK, HLA-DRB1, FCγ RIIb, IRF5, STAT4, TNFAIP3, and TNFSF13) and found that patients with SLE carrying more than 10 risk alleles demonstrated a greater risk of neurological symptoms compared with those carrying fewer than 10 risk alleles. Guerra, et al5 reported that the integrin alpha M (ITGAM) and FcγR genes contributed to apoptosis, which may lead to programmed cell death in neurons and result in NP symptoms in patients with SLE. Yang, et al14 found that ITGAM was associated with severe manifestations of SLE. May, et al15 proposed that tumor necrosis factor-α (TNF-α) causes local damage in the brain and results in demyelination.
Metaanalyses are useful because they combine multiple studies on the same alleles of genes to enhance the statistical power of the analysis and derive more reliable results of the genetic effects16,17. Previous metaanalyses studying the relationship between gene polymorphisms and SLE found that the MBL variants are risk factors for SLE, including exon 1 codon, 54 B, promoter −550L, and promoter −221X18, TNF-α –308 A/G polymorphism19, DD (deletion/deletion) genotype of intron 16 angiotensin-converting enzyme (ACE) gene20, cytotoxic T-lymphocyte antigen 4 (CTLA-4) 1722T/C polymorphism21, TNF ligand superfamily member 4 (TNFSF 4) rs2205960 polymorphism17, IL-1A –889 C/T polymorphism22, and IL-6 -174 G/C and -572 G/C polymorphisms23. Supplementary Table 1 (available online at jrheum.org) summarizes the pathological roles of specific genotypes in SLE.
SLE is a multisystem disease with diverse and protean clinical features, and genetic studies focusing on specific endophenotypes may result in reduced genetic heterogeneity24. The association between genetic polymorphism and susceptibility to NPSLE was not explored in previous metaanalyses. We propose that a metaanalysis of case-control association studies may identify candidate genes for NPSLE susceptibility. The primary aim of our metaanalysis was to assess the association between different genotypes and the risk of developing NPSLE. To achieve this aim, we combined results from previous studies and assessed the strength of the association between particular genotypes and the OR to develop NPSLE. The second aim of our study was to identify genes that have an influential effect on the NP manifestation of NPSLE.
MATERIALS AND METHODS
Search strategy
Studies of genotypes in SLE and NPSLE were systematically searched in the following databases from inception to February 2014: PubMed, EMBASE, BIOSIS, and ScienceDirect. The search terms that were used were interleukin-1, IL-1; interleukin-2, IL-2; interleukin-6, IL-6; interleukin-8, IL-8; interleukin-10, IL-10; interleukin-18, IL-18; interferon regulatory factor 5, IRF5; tyrosine kinase 2, TYK2; Fc receptor-like 3, FCRL3; Fc gamma receptor IIb, Fcγ receptor IIb, FcγRIIb; Fc gamma receptor IIa, Fcγ receptor IIa, FcγRIIa; Fc gamma receptor IIIa, Fcγ receptor IIIa, FcγRIIIa; Fc gamma receptor IIIb, Fcγ receptor IIIb, FcγRIIIb; TRAF1-C5; TNFSF4; toll-like receptor 9, TRL-9; tumour necrosis factor-α, TNF-α; lymphotoxin alpha, LTA; endothelial nitric oxide synthase, eNOS; PXK; BANK1; TNFAIP3; ITGAM; STAT4; programmed cell-death 1, PDCD1; mannose-binding lectin, MBL; RANTES, angiotensin converting enzyme; ACE; FAS; CTLA-4; C4; C2; C1q; human leukocyte antigen, HLA; T cell receptor, TCR; CR1; C3b; C4b; Immunoglobulin Gm; Km; Poly (ADP-ribose) polymerase, PARP; heat shock protein 70, hsp70; vitamin D; XRCC1; TREX-1; and a combination of key words for SLE: “systemic, erythematosus, lupus, SLE, and neuropsychiatric”. We also performed a manual review of reference lists from relevant studies. The search was limited to articles with English abstracts and to articles that reported data from humans.
Inclusion criteria and exclusion criteria
Studies were included if the following criteria were met: (1) studies that used the standard criteria of the ACR to diagnose SLE and NPSLE, (2) case–control studies that reported the allele frequencies of a particular gene in patients with SLE who did and did not experience NP symptoms, (3) prospective studies that reported the allele frequencies of a particular genotype in patients with SLE who did and did not develop NP symptoms at the end of the followup period, (4) the presence of NP symptoms was assessed clinically or using standardized instruments (i.e., subjective reports of NP symptoms were not acceptable for inclusion), and (5) studies that provided adequate information to calculate the effect size and the pooled R. Studies were excluded if one of the following criteria were met: (1) no report of the allele frequencies of a particular gene, or (2) animal studies.
Data extraction
We conducted a metaanalysis in line with the Meta-Analysis of Observational Studies in Epidemiology guidelines on the synthesis of observational data25. A selection of relevant publications was conducted independently by 2 researchers (OHY and CT), and any disagreements were resolved through discussions with RCH. The articles were deidentified [blinding of the title, author(s), year of publication, and journal name] before data extraction. The following information were extracted from each article and crosschecked by the second researcher: average age of the participants, the proportion of men, other moderators, and the relevant statistics describing the relationship between a particular genotype and the risk of developing NPSLE. Summary statistics, such as allele frequencies and the number of patients with SLE who were positive and negative for NP symptoms, were extracted or calculated from the original data.
Statistical analysis
All of the statistical analyses were performed with Comprehensive Meta-Analysis Version 2.0. A comparison was performed between the patients with NPSLE and SLE. The OR and 95% CI for developing NPSLE were calculated for genotype contrast. Tests of heterogeneity were conducted using the Q statistic, which is distributed as a chi-square variate under the assumption of the homogeneity of effect sizes. The between-study heterogeneity was assessed with the I2 statistic and p value. Random-effects model was used when there was significant between-study heterogeneity. Fixed-effects model was chosen to calculate the average OR across the relevant studies when heterogeneity was not statistically significant because the random-effects model assumes a markedly conservative null-hypothesis model in genetic metaanalysis26. Finally, Egger’s regression test was performed to test for evidence of publication bias.
RESULTS
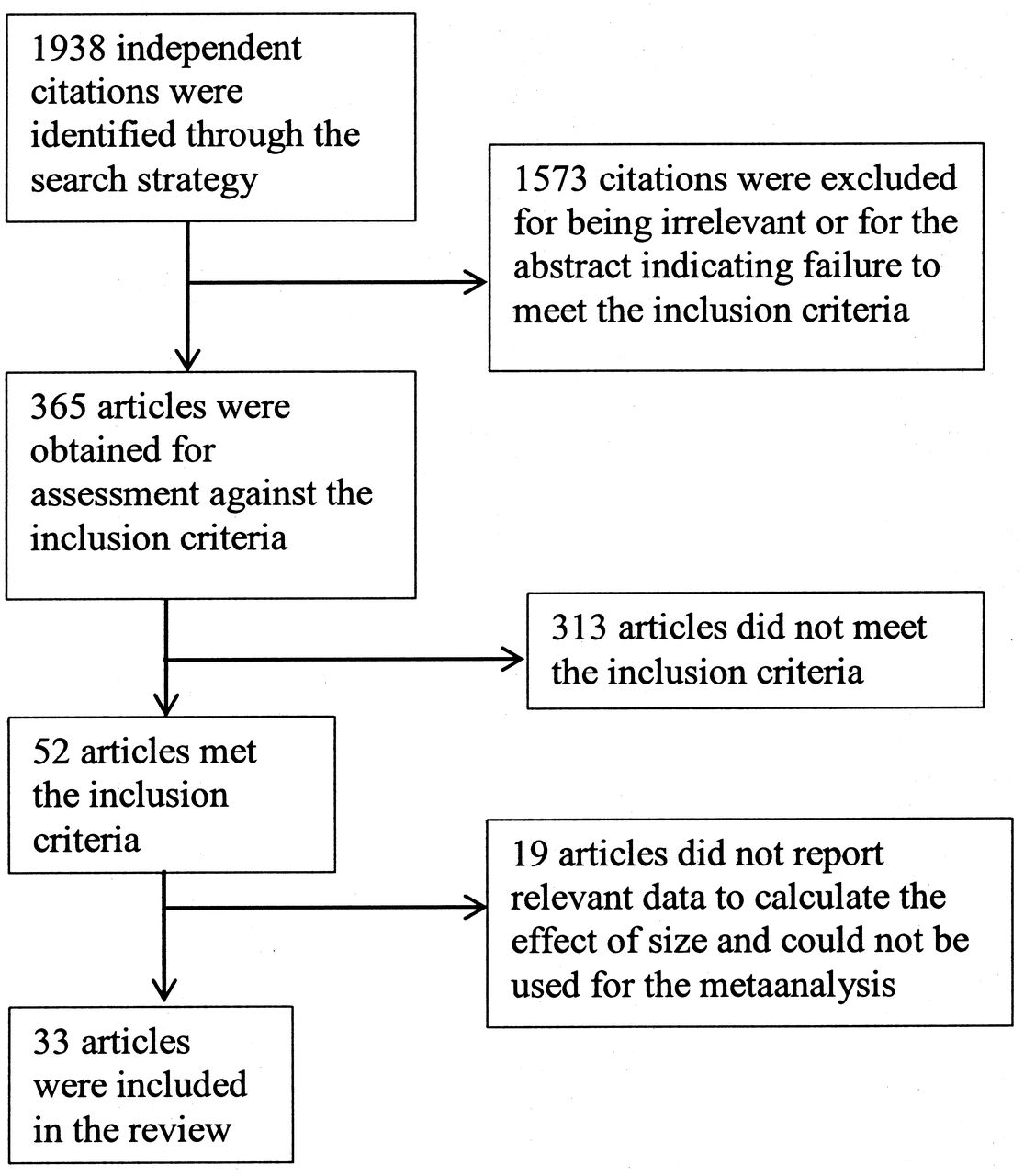
Among an initial 1938 potentially relevant articles, 52 articles met our inclusion criteria, of which 19 did not report relevant data to calculate the effect size. Finally, we included 33 articles with 905 patients with NPSLE and 4928 patients with SLE without NP symptoms in our analysis (Figure 1). Table 1 summarizes the characteristics of the included studies for our metaanalysis.

Flowchart describing the process of study selection.
Characteristics of the included studies for this metaanalysis.
A summary of the metaanalysis findings on the relationship between all of the gene polymorphisms and NPSLE is given in Table 2.
Results of the metaanalysis for genetic variants and NPSLE association.
FCγR IIIa gene polymorphism and NPSLE association
When the FF homozygotes were contrasted with VV and FV combined, there was a significant association between the homozygous FF genotype and the risk of NPSLE (OR 1.89, 95% CI 1.08–3.30, p = 0.03; Figure 2A), with no evidence of significant between-study heterogeneity (I2 = 0, p = 0.61).

Forest plots describing the metaanalysis of the association between (A) the FCγR IIIa 158 V/F polymorphism (FF vs VV + FV), (B) FCγR IIIb NA 1/2 polymorphism (NA 1/2 vs NA 1/1), and (C) FCγR IIIb NA 1/2 polymorphism (NA 1/2 vs NA 1/1+NA 2/2) and the risk of neuropsychiatric systemic lupus erythematosus. FCγR: γ Fc region.
FCγR IIIb gene polymorphism and NPSLE association
When NA1/2 was contrasted with NA1/1, a significant association between the heterozygous NA1/2 genotype and the risk of NPSLE was found (OR 2.14, 95% CI 1.07–4.30, p = 0.03; Figure 2B), with no evidence of significant between-study heterogeneity (I2 = 52.38, p = 0.15). When NA1/2 was contrasted with NA1/1 and NA2/2 combined, a significant association between the heterozygous NA1/2 genotype and the risk of NPSLE was found (OR 1.81, 95% CI 1.02–3.20, p = 0.04; Figure 2C), with no evidence of significant between-study heterogeneity (I2 = 29.10, p = 0.24).
ITGAM gene polymorphism and NPSLE association
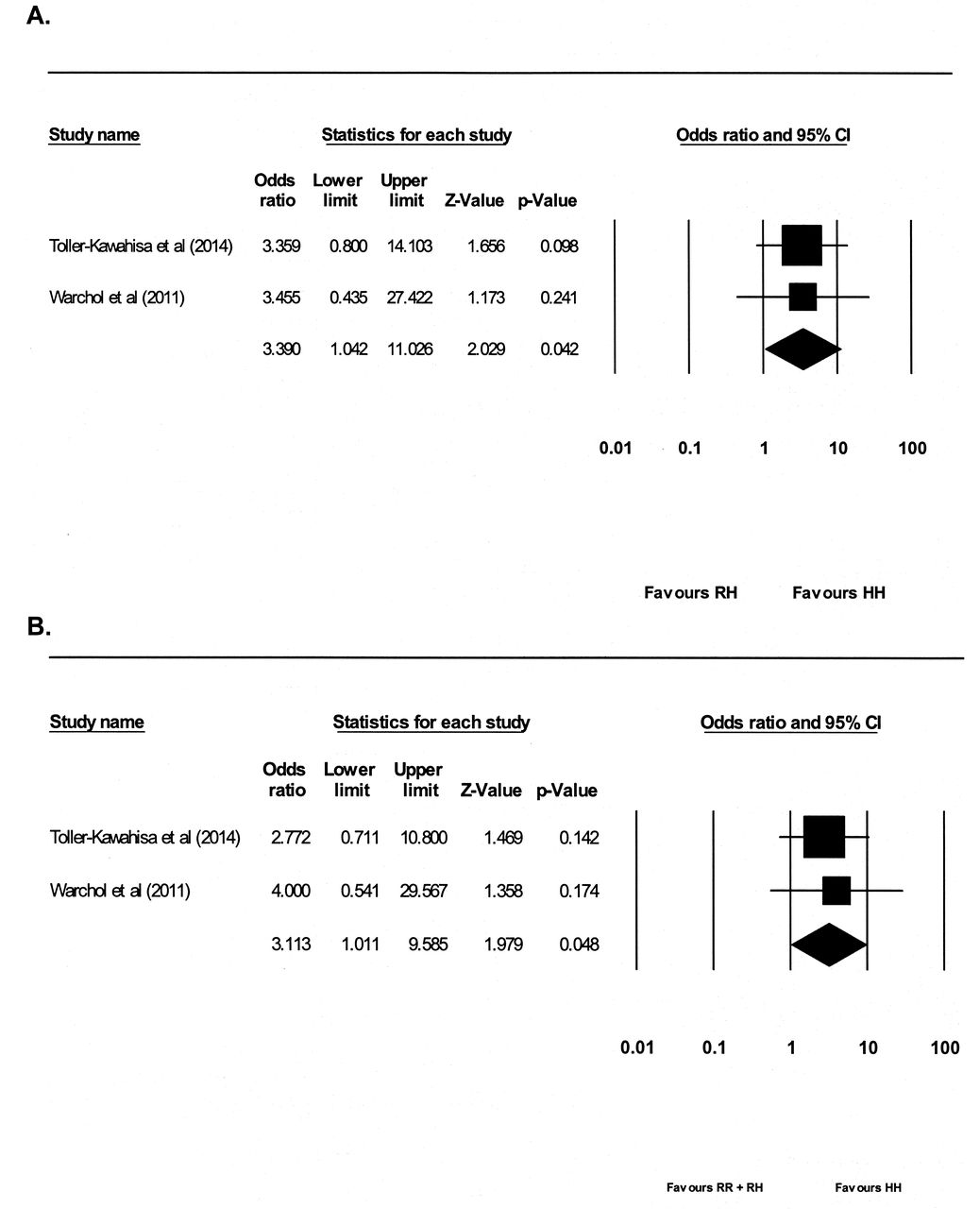
When HH was contrasted with RH, a significant association between the homozygous HH genotype and the risk of NPSLE was found (OR 3.39, 95% CI 1.04–11.03, p = 0.04; Figure 3A), with no evidence of significant between-study heterogeneity (I2 = 0, p = 0.98). When HH was contrasted with RR and HH combined, a significant association between the homozygous HH genotype and risk of NPSLE was found (OR 3.11, 95% CI 1.01–9.59, p = 0.048; Figure 3B), with no evidence of significant between-study heterogeneity (I2 = 0, p = 0.77).

Forest plot describing the metaanalysis of the association between the ITGAM rs1143679 HR polymorphism (HH vs RR + RH) and the risk of NPSLE. (A) HH versus RH, and (B) HH verus RR + RH. ITGAM: integrin alpha M; NPSLE: neuropsychiatric systemic lupus erythematosus.
Negative findings in the gene polymorphisms and NPSLE association
We did not find any association between NPSLE and MBL2 rs7096206, MBL2 Exon1, LTA 252, IL-1β -511, IL-1 RN, IL-6 -174, IL-10 promoter haplotypes (-1082 GA, -819CT, -592CA), and vitamin D receptor Bsml gene polymorphisms (p > 0.05).
Heterogeneity and publication bias
There was no statistically significant heterogeneity between studies. The I2 value varied from 0% to 73.90%, and the p value varied from 0.05 to 0.95. There was evidence of publication bias in 2 analyses (TNF-α A-308G AA vs AG, Egger’s regression coefficient = −12.12, p = 0.03; IL-6 -174GC GG+GC vs CC, Egger’s regression coefficient = 0.93, p = 0.0068). Statistically, it was not possible to assess the publication bias for analyses that involved fewer than 3 studies.
DISCUSSION
To our knowledge, ours is the first metaanalysis investigating the association between various genetic polymorphisms and NPSLE. Our metaanalysis demonstrated that the homozygous FcγRIIIa 158FF genotype, the heterozygous Fcγ IIIb NA1/2 genotype, and the homozygous ITGAM HH genotype may be associated with the susceptibility to NPSLE. The overall homozygosity for the HH genotype of the ITGAM rs1143679 gene demonstrated the highest risk for NPSLE compared with the heterozygous RH genotype, with a 3.4-fold greater risk for the development of NPSLE. Our metaanalysis highlights several important unresolved issues in the genetics of NPSLE. Future studies of NPSLE may focus on the above genes, which are related to pathways of the immune complex clearance. Our metaanalysis demonstrated no significant association of the TNF-α, MBL-2 rs7096206, MBL-Exon 1, LTA 252 rs909253, IL-6 -174GC, IL1β -511CT, and IL-1 RN polymorphisms with NPSLE. In our metaanalysis, the between-study heterogeneity was low and nonsignificant. There was no publication bias in the significant findings (i.e., the Fcγ RIIIa, Fcγ RIIIb, and ITGAM rs1143679 genotypes).
FCγRIII
Fcγ receptors are found in the IgG membrane and form an important link between the cellular and humoral elements of the immune system57. Kimberly, et al58 detected a defective Fcγ receptor-mediated uptake of IgG ligand-coated erythrocytes by macrophages in patients with SLE and postulated that the Fcγ receptor-mediated clearance defect is caused by underlying genetic etiology. Genetic polymorphisms affect the structure and function of Fcγ receptors. There are 3 different isoforms of Fcγ receptors (Fcγ RI, Fcγ RII, and Fcγ RIII), and each isoform has its own cell-anchoring mechanisms and allelic variations. Defective Fcγ receptor-mediated clearance leads to the subsequent deposition of immune complexes and contributes to the pathogenesis of SLE59. The gene of Fcγ RIII is located on chromosome 1q23. For the γγ RIIIa-V/F 158 polymorphism, F is the low-binding allele and V is the high-binding allele. The homozygosity of low-affinity FF genotype is associated with defective binding of immune complexes, increased tissue deposition, and accelerated organ damage60. VV homozygotes are more likely to bind to IG1 and IG3 compared with VF heterozygotes and FF homozygotes61. Li, et al62 performed a metaanalysis on the involvement of the Fcγ receptor IIIA –V/F 158 polymorphism in SLE and found that the homozygosity of FF genotype was associated with lupus nephritis and SLE. Our findings suggest that the homozygosity of FF genotype is associated with NP symptoms.
The Fcγ IIIb receptor is a low-affinity receptor that is expressed in neutrophils and that removes small immune complexes from the circulation. There are bi-allelic neutrophil-specific antigens (NA1 and NA2) in the Fcγ IIIb receptor gene. The NA1 allele is associated with autoimmune neutropenia63, and the NA2 allele is associated with a reduction in the capacity for phagocytosis64. There are significant ethnic differences in allele frequencies among patients with SLE. NA1 is the prominent allele in Chinese and Malay patients65, while NA2 is the prominent allele in Indian patients57. The FcγRIIIb NA1/NA2 heterozygote demonstrates a reduction in the affinity for its ligand, a higher risk of encapsulated bacterial infections, and association with lupus nephritis in Japanese patients and with endstage renal disease in Chinese patients51,66,67.
ITGAM
CD11b-integrin (ITGAM) is a member of the immune-complex processing pathway that enhances the adherence of neutrophils and monocytes to stimulated endothelium in the phagocytosis of complement cleavage fragment particles. ITGAM encodes integrin-αM (CD11b+), which is a molecule that combines with integrin-β2 to form a leukocyte-specific integrin14. The αMβ2-integrin forms cell surface receptors on monocytes and neutrophils to bind intracellular adhesion molecule 168. The ITGAM gene is associated with SLE susceptibility in Europeans69, Hong Kong Chinese, and Thai14. The ITGAM genotype polymorphism was proposed to be one of the strongest genetic risk factors70 and predicts disease manifestations in SLE35. The rs1143679 variant of the ITGAM gene encodes an arginine-to-histidine amino acid change at position 77 (R77H) in the β propeller domain of CD11b. The presence of the H allele increases CR3 expression and aggravates the inflammatory process in patients with SLE by activating complements, antiphospholipid autoantibodies, and neutrophils, causing arterial and venous thrombosis and the risk of lupus nephritis71. In addition to renal disease and discoid rash72, the ITGAM genotype polymorphism may increase the risk of NP manifestations by cell-mediated immunological reactions.
Other genotypes
Our results suggest that there is no significant association of the polymorphisms of MBL2 rs7096206, exon1, LTA 252 rs909253, IL-6 -174GC, IL-1βC-511TC, and IL-1 RN with the susceptibility to develop NPSLE. IL-10 is a major immunoregulatory cytokine, and IL-10 genotypes are strongly associated with renal disease9. Rood, et al12 found that IL-10 promoter haplotypes are implicated in the pathogenesis of NPSLE, but another study in Thai patients with SLE did not support those findings34. Other studies on the IL-10 promoter genes in patients with SLE did not assess NP symptoms73,74,75,76. Further studies are required to investigate the involvement of IL-10 promoter haplotypes in NPSLE.
Our metaanalysis has inherited the limitations of the case-control genetic association studies. First, the number of studies for each genotype was small, and our metaanalysis was underpowered to detect differences in some of the genotypes between patients with NPSLE and SLE. Second, Li, et al60 found that the Fcγ RIII a-FF genotype was associated with a high risk of developing SLE in Asians and Europeans, but not in Africans. However, we were not able to perform an ethnic subgroup analysis for each genetic polymorphism as a result of the small number of published studies. Third, we were not able to address intergenetic and gene-environment interactions (e.g., smoking and ultraviolet light exposure). Fourth, NPSLE consists of a broad spectrum of very different clinical manifestations. In our metaanalysis, we were not able to study the relationship between specific NP manifestation and its association with specific allele. We hope that researchers will attempt to study the association between specific NP symptom and genotype in the future. Finally, replication in multiple studies is essential for the validation of genetic associations, and metaanalysis, which combines results across multiple samples from independent studies, is a key component for genetic findings. The reliability of results is not reduced when a metaanalysis pools summary statistics from individual studies; however, unavailability of individual-level data and technical and statistical differences limit the available options for multiple-testing adjustments. Thus, our study used sample-size–weighted meta statistics, which is a common approach for combining results in metaanalysis50.
Our metaanalysis serves as a preliminary overview of the involvement of various genes as a potential etiological factor in NPSLE and sheds light on the pathogenesis of NPSLE. Our results also provide a foundation for future genetics studies to focus on endophenotypes. Boackle77 proposed the use of clinical features such as NP status to classify patients with SLE and to reduce genetic heterogeneity. The findings in our metaanalysis require further replication. Further longitudinal studies with larger sample sizes and different ethnicities are required to study the interaction between the Fcγ RIIIa, Fcγ RIIIb, and ITGAM genotypes in the causation of NPSLE. With the publication of more genetic studies in NPSLE, a future metaanalysis should attempt to combine the cumulative number of risk alleles and to assess the risk of developing NP complications in patients with SLE.
Our metaanalysis suggests that polymorphisms in the pathways of immune complex clearance might contribute to the susceptibility of NPSLE. The homozygous FcγR IIIa -158 FF genotype, the heterozygous FcγR IIIb NA1/2 genotype, and the homozygous ITGAMHH genotype were associated with NPSLE. The defects in the immune complex clearance pathway may shed light on new therapeutic targets for NPSLE. Further genetic studies focusing on the NP endophenotype with larger sample sizes are required to replicate the findings of our metaanalysis.
ONLINE SUPPLEMENT
Supplementary data for this article are available online at jrheum.org.
- Accepted for publication October 15, 2015.








{kind=link}
{kind=link}
{kind=link}