

Abstract
Objective. Rheumatoid arthritis (RA) that is negative for anticitrullinated protein antibodies (ACPA) is a subentity of RA, characterized by less severe disease. At the individual level, however, considerable differences in the severity of joint destruction occur. We performed a study on genetic factors underlying the differences in joint destruction in ACPA-negative patients.
Methods. A genome-wide association study was done with 262 ACPA-negative patients with early RA included in the Leiden Early Arthritis Clinic and related to radiographic joint destruction over 7 years. Significant single-nucleotide polymorphisms (SNP) were evaluated for association with progression of radiographic joint destruction in 253 ACPA-negative patients with early RA included in the Better Anti-Rheumatic Farmaco Therapy (BARFOT) study. According to the Bonferroni correction of the number of tested SNP, the threshold for significance was p < 2 × 10−7 in phase 1 and 0.0045 in phase 2. In both cohorts, joint destruction was measured by Sharp/van der Heijde method with good reproducibility.
Results. Thirty-three SNP associated with severity of joint destruction (p < 2 × 10−7) in phase 1. In phase 2, rs2833522 (p = 0.0049) showed borderline significance. A combined analysis of both the Leiden and BARFOT datasets of rs2833522 confirmed this association with joint destruction (p = 3.57 × 10−9); the minor allele (A) associated with more severe damage (for instance, after 7 yrs followup, patients carrying AA had 1.22 times more joint damage compared to patients carrying AG and 1.50 times more joint damage than patients carrying GG). In silico analysis using the ENCODE and Ensembl databases showed presence of H3K4me3 histone mark, transcription factors, and long noncoding RNA in the region of rs2833522, an intergenic SNP located between HUNK and SCAF4.
Conclusion. Rs2833522 might be associated with the severity of joint destruction in ACPA-negative RA.
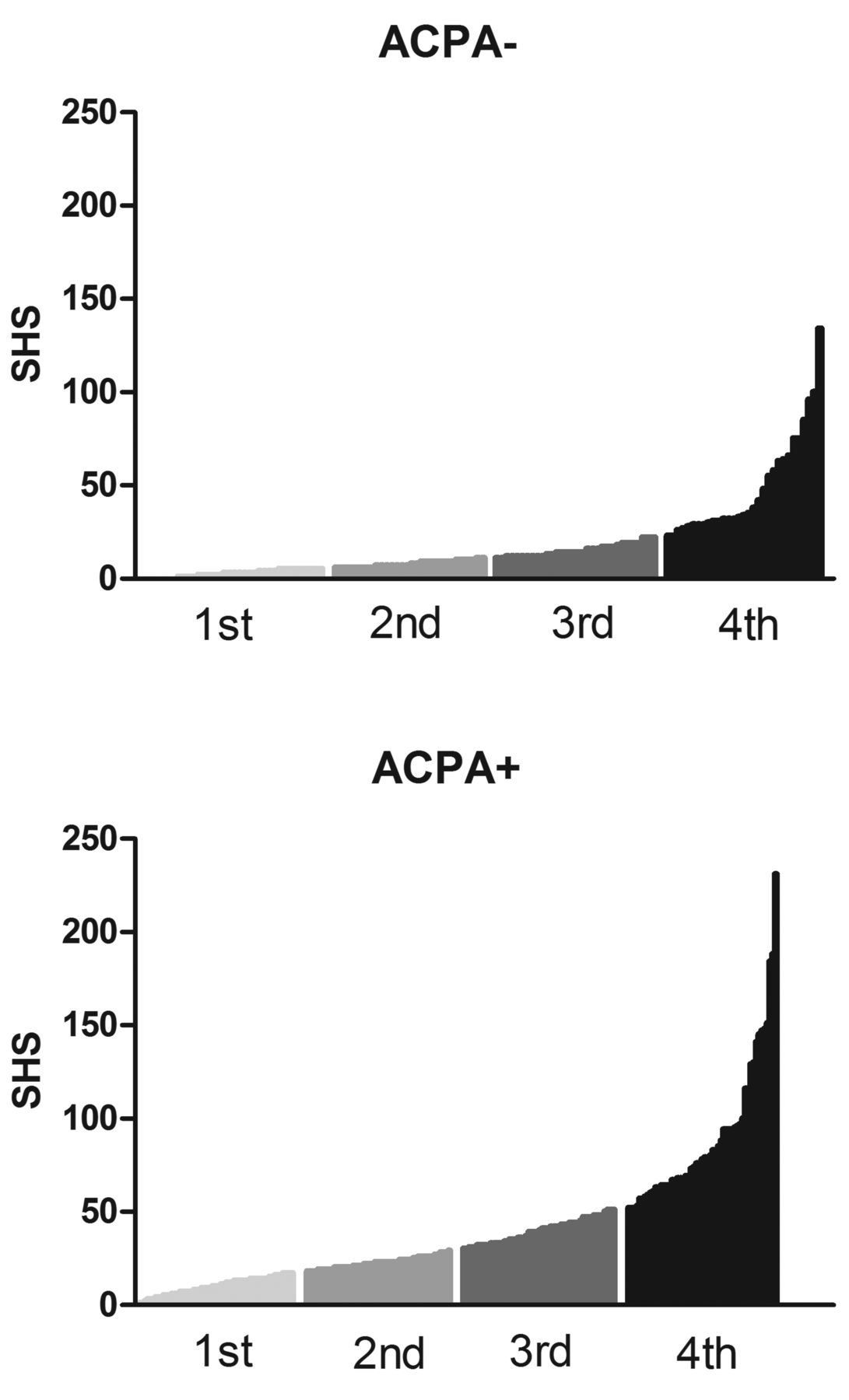
Rheumatoid arthritis (RA) is diagnosed by clinical characteristics and not based on markers that are directly reflecting the underlying pathophysiological processes. Nonetheless, studies have found differences in genetic susceptibility factors, histopathology, and outcome of RA patients with or without anticitrullinated protein antibodies (ACPA)1,2,3. Therefore RA is considered to consist of 2 subentities, 1 characterized by the presence of ACPA and another without these autoantibodies. On the group level, ACPA-negative patients with RA have a less severe disease course with less radiographic joint damage [measured by the Sharp/van der Heijde method (SvdH)] than do ACPA-positive patients. On the individual level, however, disease severity is variable between patients, and severe joint destruction also occurs in autoantibody-negative patients (Figure 1). The mechanisms driving the interindividual differences in the severity of joint destruction in ACPA-negative RA are largely unknown. Genetic factors likely play a role. However, few genetic studies have focused on radiological progression in ACPA-negative RA. The majority of studies examined the total group of patients with RA or included predominantly ACPA-positive patients with RA.

Sharp/van der Heijde scores (SHS) after 5 years of followup in ACPA– and ACPA+ patients included in the Leiden Early Arthritis Cohort. Depicted are the first, second, third, and fourth quartiles of patients and their SHS scores after 5 years of followup. ACPA: anticitrullinated protein antibodies.
The heritability of joint damage measured by SvdH in the total population of patients with RA is estimated to be 45–58%4. Whether this is different for ACPA-positive and ACPA-negative RA is unclear. Joint destruction in RA is considered the net result of the influence of inflammation on bone. Although hypothetically several processes are similar in ACPA-positive and ACPA-negative RA (for instance, processes that determine the sensitivity of bone and cartilage to destruction in response to inflammatory stimuli), other pathways (such as immunological mechanisms) may be differently regulated in ACPA-positive and ACPA-negative RA and affect the severity of joint damage. The assumption that some risk factors for progression of joint destruction are shared between these subsets of RA and others are subset-specific is in line with findings in the field of the genetics of RA susceptibility. It has also been shown that some genetic risk factors predispose to both ACPA-positive and ACPA-negative RA, whereas other genetic factors predispose to only 1 of these 2 subsets5.
In our study, we aimed to identify genetic factors associated with severity of joint damage in ACPA-negative RA. We performed a 2-stage study in white patients, starting with a genome-wide screen. Both stages included cohorts of early ACPA-negative patients with RA who had serial radiographs over time and were recruited in periods when treatment strategies were milder than today and when biological therapy was uncommon. Both longitudinal observational cohorts were unique with regard to the number of ACPA patients who had radiographs taken.
MATERIALS AND METHODS
Patients
In both phases of our study, RA was defined according to the 1987 American College of Rheumatology criteria. All patients gave written informed consent. The medical ethical committees of the participating centers (in Leiden and in Sweden) approved the study.
In the first phase, 276 ACPA-negative patients with early RA who were included in the Leiden Early Arthritis Cohort between 1993 and 2006 were studied6. ACPA was measured in stored sera that were collected at the first visit using ELISA (Immunoscan RA Mark 2, Eurodiagnostica). Samples with a value < 25 units/ml were considered negative according to the manufacturer’s instructions. Radiographs of the hands and feet were taken at baseline and yearly thereafter during 7 years of followup (in total, 1266 sets of hands and feet radiographs). Radiographs were chronologically scored by 1 reader using SvdH with good ICC (intraclass observer correlation coefficient)7. To calculate this ICC, several radiographs were blindly scored twice by the same reader, and agreement was calculated. All ICC were > 0.8, the threshold considered to reflect good scoring of radiographs. Strategies were different in 3 treatment periods (1993–1995, initial treatment with nonsteroidal anti-inflammatory drugs; 1996–1998, initial treatment with chloroquine or sulfasalazine; and 1999–2006, prompt treatment with methotrexate or sulfasalazine), as described in more detail6.
The second phase concerned 253 ACPA-negative patients with RA included in the Better Anti-Rheumatic Farmaco Therapy (BARFOT) cohort, a Swedish multicenter observational study of patients with early RA (disease duration ≤ 1 yr)8. Clinical, laboratory, and radiological assessments were performed at inclusion and after 1, 2, and 5 years of followup. Hands and feet radiographs (total number 842) were scored according to the SvdH method by 2 readers with good ICC. To calculate the between-reader ICC, some radiographs were scored by both readers. Average scores of the readers were used. During followup, 50 patients participated in a 2-year randomized study on low-dose prednisolone as an addition to disease-modifying antirheumatic drug therapy8. Baseline characteristics of the ACPA-negative study populations are shown in Table 1.
Baseline characteristics of the ACPA-negative study population.
Genotyping
In phase 1, genotyping was done using Illumina Human CytoSNP-12v2, as described9. Results of 244,655 SNP were obtained. Of these, all call rates were > 97% and no SNP were excluded because of a low call rate. Thirty-one SNP were excluded from the analyses because they were not in Hardy-Weinberg equilibrium (threshold 10-4), leaving 244,624 SNP to be analyzed for an association with the progression of joint destruction. DNA on 276 patients was used for genotyping; 2 patients were excluded because of failed genotyping, 1 patient because of high homozygosity, and 11 patients because they were relatives (based on identity-by-state analysis). Thus, 244,624 SNP were studied in 262 ACPA-negative patients. No patient was removed because of population stratification.
In phase 2, SNP that showed a significant association in the first phase and with minor allele frequency (MAF) > 0.1 (n = 18) were genotyped using Sequenom iPLEX. Because the number of radiographs in the second dataset was lower than in the first dataset, yielding a lower power in dataset 2 compared to dataset 110, from the 33 variants identified in phase 1, only the SNP that had a MAF of > 0.1 (n = 8) were studied for an association with joint destruction in ACPA-negative patients included in the BARFOT cohort. In this way, we tried to lower the chance of obtaining false-positive results. The power to find statistically significant differences for SNP with a lower MAF was estimated to be insufficient. Apart from rs4926674, for which genotyping failed, success rates were all > 98.5% and error rates 0% (based on the samples typed in duplicate). No patients were excluded from the analyses.
Statistical methods
In phase 1, associations with the SNP with MAF > 0.05 and radiographic joint destruction were studied based on a linear mixed effects model for the longitudinal log-transformed SvdH data11. A detailed description of the statistical model used is available from the authors on request.
In phase 2, the same model as described in phase 1 was used. Also here, analyses were corrected for age, sex, and treatment differences (participation in the corticosteroid trial). The Bonferroni correction for the number of uncorrelated SNP (R2 = 0.8, n = 11) was used; the threshold for significance was 0.0045.
The results obtained in the 2 cohorts for the strongest associating SNP were combined into 1 analysis. This combined analysis was performed by combining the data of the 2 cohorts and correcting for participating in either the Swedish or the Dutch cohort in a statistical analysis similar to the one described in phase 1 and 2; so a linear mixed-effects model was applied on all data. This combined analysis bears similarities to the fixed-effects metaanalysis in the sense that both studies are estimating the same effect size, and the contribution of each study on the estimation of the combined effect is determined by the amount of information available to each one.
All analyses were done using the R statistical software package12. The linear mixed effects model was fitted in R using the function lme from the R package nlme, and the nonlinear time evolutions were modeled using function ns from the R package splines13,14.
To further investigate potential sample contaminations, swaps, and duplications as well as pedigree errors and unknown familial relationships, we computed the pairwise IBD (identity by descent) to find pairs of individuals who look too similar to each other, i.e., more than we would expect by chance in a random sample. These computations were done with PLINK15. We then applied classic multidimensional scaling (or principal coordinates analysis)16 on the 2-dimensional distances between the points in the IBS (identity by state) matrix. The mapping of the patients on the space of the first 2 principal components is used to identify potential genetic outliers. For this analysis we have used the R package snpMatrix12.
In silico analysis of the rs2833522 region
The region of rs2833522 was analyzed for DNA and histone modifications, the presence of DNA regulatory elements (including enhancers and transcription factor binding), and for identification of coding and noncoding RNA transcripts, focusing on cell types relevant for RA, using The Encyclopedia of DNA Elements [ENCODE - University of California at Santa Cruz (UCSC) Genome Browser GRCh37/hg1917] and Ensembl 2014 databases18. An annotation data file in BED format, specifying the location of the long-coding lncRNA AP000255.6-001, as defined in Ensembl, was uploaded in the UCSC Genome Browser and is available from the authors on request, as a custom annotation track AP000255.6-001.
RESULTS
Phase 1
In total, 244,624 SNP were studied in 1266 sets of hands and feet radiographs of 262 ACPA-negative patients in relation to the radiological severity of joint destruction over 7 years of followup. A Q-Q plot for observed versus expected values of the likelihood ratio test statistic under a chi-squared distribution with 4 degrees of freedom is shown in Supplementary Figure 1, available from the authors on request. The lambda score for genomic control was 0.994. The results are depicted in the Manhattan plot (Figure 2). Thirty-three SNP related significantly (p < 2 × 10−7) to the progression of joint destruction (Table 2).

Manhattan plot of all p values obtained in phase 1.
SNP that passed the significance threshold of 2 × 10−7 in phase 1 and the p value obtained in phase 2.
Phase 2
Two SNP associated with the progression of joint destruction: rs2833522 (p = 0.0049) and rs17763915 (p = 0.047). However, neither passed the Bonferroni threshold for multiple testing, although the p value of rs2833522 was just above this threshold. The joint destruction scores of the ACPA-negative Leiden and BARFOT patients having a minor, heterozygous, or major genotype of this SNP are depicted in Figures 3A and 3C. The fitted profiles by the model of both cohorts are presented as well (Figures 3B and 3D). As shown, in both cohorts presence of the minor allele was associated with more severe damage progression. After 7 years of followup, patients carrying the AA genotype had a 1.22 times higher estimated joint damage score as predicted by our statistical model (estimated log SvdH + 1) compared to patients carrying the AG genotype and a 1.50 times higher estimated joint damage score compared to the patients carrying the GG genotype. Supplementary Table 1, available from the authors on request, gives the effect sizes estimated for the other timepoints studied.

Progression of joint damage for the ACPA-negative patients with RA carrying the homozygous major, heterozygous, and homozygous minor genotype of rs2833522. Portrayed are the sample mean SHS scores, thus the raw SHS data on the normal scale (A, Leiden EAC; C, BARFOT), the mean log-transformed SHS as modeled by the linear mixed model (B, Leiden EAC; D, BARFOT), and the mean log-transformed SHS as modeled by combined analysis of both cohorts (E). ACPA: anticitrullinated protein antibodies; RA: rheumatoid arthritis; SHS: Sharp/van der Heijde scores; EAC: Early Arthritis Cohort; BARFOT: Better Anti-Rheumatic Farmaco Therapy study.
Combined analysis
Finally, the data of rs2833522 of both cohorts were analyzed in a combined analysis. This showed an association (p = 3.57 × 10−9); the fitted profiles are depicted in Figure 3E.
Results of in silico analysis of the rs2833522 region
Rs2833522 is located on chromosome 21, between SFRS15, also known as SCAF4, and HUNK (hormonally upregulated new-associated kinase)19. A new lncRNA, AP000255.6-001 (indicated as green custom annotation track, Supplementary Figure 1A, available from the authors on request), has been identified between SCAF4 and rs2833522 (70 kb from rs2833522)16. Additionally, several genes are predicted by N-SCAN20,21, Genscan22, and H-Inv 7.023,24,25,26 gene predictions to map within the rs2833522 region, with the introns of some spanning rs2833522 (Supplementary Figure 1A, available from the authors on request). Further, in peripheral blood mononuclear cells (PBMC), rs2833522 co-localizes with H3K4me3 histone mark (ENCODE: GEO Sample Accession GSM788075), associated with active chromatin architecture, while in K-562 leukemic cells a repressive H3K27me3 histone mark is present ∼200 bp downstream rs2833522 (ENCODE: GSM788088; Supplementary Figure 1B, available from the authors on request). Some transcription factors (ENCODE: GSM777644, GSM777641, GSM777640, GSM777637), including T cell acute lymphocytic leukemia [TAL1; Transcription Factor ChIP-seq V4 (161 factors) with Factorbook repository motifs from ENCODE], were shown to localize in the close proximity of rs2833522 (Supplementary Figure 1B, available from the authors on request) in K-562 cells.
DISCUSSION
Not many genetic studies in RA have focused on the ACPA-negative subset of the disease, neither with regard to RA susceptibility nor to RA severity. Genome-wide association studies (GWAS) have identified > 100 genetic risk factors for RA development27,28,29,30, but the majority of patients in these studies was ACPA-positive. A recent GWAS compared ACPA-negative RA patients with controls and identified a peak in the HLA region and suggestive evidence for an association in the CLYBL locus; this locus was not identified in the studies on the total group of patients with RA or ACPA-positive RA9. Additionally, a study using the Immunochip custom SNP array performed stratified analyses in ACPA-positive and ACPA-negative patients and observed 2 variants predisposing to ACPA-negative RA with a genome-wide significant p value [1 variant in the HLA region (rs414332) and in another variant in ANKRD55 (rs71624119)]5. Together these findings support the notion that there are differences in the genetic variants predisposing to ACPA-positive and ACPA-negative RA. A possible reason for the scarcity of genetic studies on ACPA-negative patients with RA is the fear of phenotypic misclassification. ACPA-negative RA is sometimes thought to be heterogeneous and containing patients with unrecognized diseases other than RA31,32. HLA is not a risk factor for severity of disease within ACPA-positive or ACPA-negative disease33,34. A study attempted to address this issue and evaluated a broad range of characteristics (clinical, serological, and radiological) at first presentation and during the course of disease, using different variable reduction techniques. Based on the characteristics studied, no subgroups of patients could be discerned35, and it was concluded that for risk-factor studies, ACPA-negative patients with RA can be studied as 1 group.
There are also few studies focusing on the radiographic progression of RA within the ACPA-negative subset of the disease. Some of the ACPA-negative patients with RA, however, have severe radiographic progression (Figure 1), and the factors or processes underlying these interindividual differences are unknown. Because evaluation of genetic variants may lead to new insights into the pathophysiology of radiographic progression in this disease subset, we performed the first (to our knowledge) GWAS on the severity of joint destruction in 2 cohorts of white ACPA-negative patients with RA. In our 2-staged study, 33 SNP passed the threshold of genome-wide significance in phase 1, and 2 of those were associated with the severity of joint damage in phase 2 at the 5% significance level. After the rather conservative Bonferroni correction for multiple testing in phase 2, rs2833522 showed borderline statistical significance (p = 0.0049, whereas the Bonferroni threshold for significance is p = 0.0045).
The directionality of the effect was similar in both cohorts: presence of the minor allele of rs2833522 was associated with more severe joint destruction. Rs2833522 is an intergenic SNP, located between HUNK19 and SCAF4 on chromosome 21. SCAF4 is an RNA-binding protein, expressed in blood mononuclear cells that may physically and functionally link transcription and pre-mRNA processing. Neither HUNK nor SFRS15 have been associated with autoimmune diseases. Similar to many other disease-linked intergenic variants that have been identified in GWAS, it is complicated to trace the mechanism underlying the observed associations. In silico analysis of SNP regions for chromatin architecture, DNA regulatory elements, and (non)coding RNA transcripts may indicate whether the identified genetic region is differentially expressed in relation to the genotype36. For rs2833522, there is H3K4me3 histone mark overlapping the SNP in PBMC. Also, some transcription factors, including TAL1, are localized near the SNP; TAL1 was recently described as a regulator of osteoclast differentiation37. Further, a new lncRNA AP000255.6-001 has been discovered and several other genes are predicted in the ENCODE database to reside within the SNP region38. Further experimental studies on chromatin architecture and noncoding RNA transcripts in this region are required to formally validate these downstream effects in the cell types participating in RA. Such studies might increase the understanding of how rs283522, or another linked SNP in this locus, affects the progression of joint damage within patients with ACPA-negative RA.
A proportion of the ACPA-negative patients studied were rheumatoid factor (RF)-positive (24%). A stratified analysis of rs2833522 in ACPA-negative patients with RA who were positive (first group) and negative (second group) for RF yielded significant results in both groups (p = 0.015 and p = 0.011, respectively), indicating that the association observed was not driven by the presence of RF in some of the patients.
In phase 2 an association was observed for rs17763915, which is located 257821 kb from the SLC4A4, encoding for a sodium bicarbonate cotransporter. To our knowledge, this gene has thus far not been related to RA or other autoimmune or antiinflammatory disorders. Because this association disappeared after correction for multiple testing, we cannot make a definite conclusion on the value of this SNP for joint damage severity.
A genome-wide approach is focused on identifying true positive risk factors and the presence of false-negative results is inherent to the study design. In our case, the risk of false negatives is of special importance. The Bonferroni correction for multiple testing applied in phase 1 is rather conservative, especially because we corrected for the total number of SNP and not the number of uncorrelated SNP; this choice may have introduced false negatives. Further, a disadvantage of the array that was used in phase 1 was that it covered relatively few variants, making it likely that not all genetic variants associated with joint damage severity in ACPA-negative RA were tested. To lower the risk of obtaining false-positive associations, we decided not to perform SNP imputation using the 1000 Genome Project dataset. Nonetheless, the choice of the array and the risk of false-negative findings do not affect the validity of the positive results we obtained.
Another issue in studies on radiographic progression in general and radiographic progression in ACPA-negative RA in particular is the statistical power. The number of patients with longitudinal disease outcome data that was also collected in the era when treatment was less intensive than it is today is relatively low. We studied cohorts with ACPA-negative patients, which reduced the available numbers of patients and radiographs even more. Our study did not include a third phase because we did not identify a longitudinal observational cohort with ACPA-negative patients with RA that included an equal or larger number of radiographs compared to the number of radiographs studied in phases 1 and 2. Cohorts of ACPA-negative patients with RA who have repeated radiographic measurements available are extremely rare. Another issue was the severity of joint damage, which was relatively mild in a large number of ACPA-negative patients with RA, and the differences in progression rates between genotype groups were therefore lower than those observed within the total population of patients with RA or within the ACPA-positive population with RA. The relatively low sample size and the low frequency of patients with severe progression decreased the ability to find significant associations. We tried to minimize the risk of false-positive findings by using 2 large cohorts in which repeated radiographic measurements were available. However, we cannot completely rule out the risk of false-positive results.
Despite these limitations, a strength of the cohorts used is that the patients were radiographed serially in time. As shown recently, repeated measurements yield more precise estimations of the radiographic progression rate and a considerably increased power10. This characteristic of the data explains the ability to identify variants passing the Bonferroni cutoff for multiple testing correction in phase 1. Further, the statistical model used in this study took advantage of the within-patient correlations present in repeated radiological measurements. This model was chosen because, especially in ACPA-negative patients, the rate of joint destruction is not homogeneous through the whole followup period and differences between the patients’ joint damage progression profiles are present. The adoption of spline functions into the linear mixed-effects model allowed more precise modeling of the progression of joint damage over the total followup period. Despite the advantage of being able to flexibly model the nonlinear profiles in time, a disadvantage of the chosen model is that we cannot quantify the SNP effect over the whole followup period using an overall effect size. Therefore, to visualize the SNP effects on the progression of joint destruction, we presented the raw SvdH data and the fitted marginal profiles in time per genotype category (Figure 3).
Another strength of the 2 observational cohorts that were studied was the similarity in design and populations. Both cohorts consisted of adult, Western European patients with early RA and in both cohorts treatment was much less intense than today. Both cohorts were scored according to the SvdH method with high reproducibility. The absolute values in SvdH were slightly different between the cohorts, perhaps because the readers of both cohorts were not trained together. This did not affect the analyses or conclusions because the relative differences in radiographic severity between genotypes were compared.
The previously identified RA susceptibility loci ANKRD555 and CLYBL9 were not in the list of variants identified in phase 1 of our study. This might indicate that the genetic variants, and hence the underlying pathophysiological processes, of developing ACPA-negative RA and progression of ACPA-negative RA are different.
We found that rs2833522 might be associated with the severity of joint damage in ACPA-negative RA. In silico analysis using ENCODE and Ensembl databases, showing marks of transcription, increased the interest in the region of rs2833522. Larger longitudinal studies are needed for final confirmation of the genetic association, and subsequent functional studies are required to elucidate the processes relevant for joint destruction in RA that are influenced by this genetic variant.
The following discrepancy has since come to our attention: ACPA typing was done in sera of > 800 patients with RA from the BARFOT cohort, among whom are the 253 ACPA-negative patients who are part of the present article; testing was done for another research project. In that project, 22 patients who were determined to be ACPA-negative in the Swedish laboratory were found ACPA-positive by ELISA done in Leiden. We therefore sought to determine whether our results were dependent on these 22 patients. However, in analyzing the data without the 22 patients, we obtained similar results and conclude that after excluding the 22 patients from our analysis, the results remain unchanged.
Acknowledgment
We acknowledge the ENCODE Consortium: NCBI RefSeq Project, Michael Brent’s Computational Genomics Group at Washington University St. Louis (N-SCAN prediction data), Aaron Tenney in the Brent lab and Robert Zimmermann, currently at Max F. Perutz Laboratories in Vienna, Austria (implementation of N-SCAN); Chris Burge (Genscan program), as well as the Biomedicinal Information Research Center, National Institute of Advanced Industrial Science and Technology, Japan; the Chinese National Human Genome Center, the Deutsches Krebsforschungszentrum, Helix Research Institute Inc., the Institute of Medical Science in the University of Tokyo, the Kazusa DNA Research Institute, the Mammalian Gene Collection, and the Full-Length Long Japan project. We acknowledge the laboratories of Peggy Farnham (University of Southern California/Norris Cancer Center; previously at the University of California at Davis) and Michael Snyder at Stanford University, who generated and analyzed the data on PBMC H3K4me3 histone modifications by ChIP-Seq Signal from ENCODE/Sydh (GEO Sample Accession: GSM788075) and K562 H3K27me3 histone modifications by ChIP-Seq Signal from ENCODE/Sydh (GSM788088); the collaboration between University of Chicago (Kevin White, Subhradip Karmakar, Nick Bild, Alina Choudhury) and Argonne National Laboratory (Marc Domanus) that created the data and annotations in K562 FOS GFP-tag TFBS Signal from ENCODE/UChicago (GSM777644), K562 GATA2 GFP-tag TFBS Signal from ENCODE/UChicago (GSM777641), K562 HDAC8 GFP-tag TFBS Signal from ENCODE/UChicago (GSM777640), and K562 NR4A1 GFP-tag TFBS Signal from ENCODE/UChicago (GSM777637). We acknowledge the Myers Laboratory at the HudsonAlpha Institute for Biotechnology and the laboratories of Mark Gerstein, Sherman Weissman at Yale University, Kevin Struhl at Harvard, Kevin White at the University of Chicago, and Vishy Iyer at the University of Texas, Austin, for the ChIP-seq data in ENCODE-Transcription Factor ChIP-seq V4 (161 factors) with Factorbook motifs from ENCODE, the ENCODE Analysis Working Group and Anshul Kundaje (processing these data into uniform peak calls), UCSC (clustering of the uniform peaks), and Jie Wang, Bong Hyun Kim, and Jiali Zhuang of the Zlab (Weng Laboratory) at UMass Medical School (the Factorbook motif identifications and localizations, assistance with interpretation).
Footnotes
Supported by the Masterswitch project and BtheCure project. The work of Dr. van der Helm-van Mil is supported by the Dutch organization for scientific research (ZonMW). The Dutch Arthritis Foundation (Reumafonds) provided financial support for the genotyping.
- Accepted for publication February 24, 2015.








{kind=link}
{kind=link}
{kind=link}