

Abstract
Objective. To describe the longterm safety and efficacy profile of tofacitinib in patients with moderate to severe active rheumatoid arthritis (RA).
Methods. Data were pooled from 2 open-label studies (NCT00413699, NCT00661661) involving patients who had participated in qualifying phase I, II, or III index studies of tofacitinib. Safety data included over 60 months of observation; efficacy data are reported up to Month 48. Treatment was initiated with tofacitinib 5 or 10 mg twice daily. Primary endpoints were adverse events (AE) and laboratory safety data. Secondary endpoints included American College of Rheumatology (ACR) response rates, and Disease Activity Score (28 joints) (DAS28)-4[erythrocyte sedimentation rate (ESR)] and Health Assessment Questionnaire-Disability Index (HAQ-DI) assessments.
Results. Overall, 4102 patients were treated for 5963 patient-years; mean (maximum) treatment duration was 531 (1844) days; 20.8% of patients discontinued treatment over 60 months. The most common AE were nasopharyngitis (12.7%) and upper respiratory tract infection (10.5%). Serious AE were reported in 15.4% of patients with an exposure-estimated incidence rate of 11.1 events/100 patient-years. Serious infections were reported in 4.5% of patients with an exposure-estimated incidence rate of 3.1 events/100 patient-years (95% CI: 2.66–3.55). Mean values for laboratory variables were stable over time and consistent with phase II and III studies. Persistent efficacy was demonstrated through Month 48, as measured by ACR response rate (ACR20/50/70) DAS28-4-ESR, and HAQ-DI. Safety and efficacy were similar for patients receiving tofacitinib as monotherapy or with background nonbiologic disease-modifying antirheumatic drugs.
Conclusion. Tofacitinib demonstrated consistent safety and persistent efficacy over 48 months in patients with RA.
Rheumatoid arthritis (RA) is a chronic and disabling autoimmune disease associated with articular inflammation and damage, functional decline, and accelerated mortality1,2.
Tofacitinib is a novel, oral Janus kinase (JAK) inhibitor for the treatment of RA. In cellular settings where JAK signal in pairs, tofacitinib preferentially inhibits signaling by heterodimers containing JAK3 and/or JAK1, with functional selectivity over receptors that signal through pairs of JAK23. Inhibition of JAK1 and JAK3 by tofacitinib blocks signaling through the common γ-chain-containing receptors for several cytokines, including interleukin 2 (IL-2), -4, -7 -9, -15, and -213. These cytokines are integral to lymphocyte activation, proliferation, and function, and inhibition of their signaling may thus result in modulation of multiple aspects of the immune response3. The relevance of specific JAK combinations to therapeutic effectiveness is unknown.
The clinical, functional, and radiographic efficacy and safety of tofacitinib dosed at 5 and 10 mg twice daily (BID) has been reported in patients with active RA in randomized phase II4,5,6,7,8 and phase III studies9,10,11,12,13,14. Full characterization of the safety profile, particularly over the longer term, is required.
Double-blind, randomized controlled trials (RCT) represent the gold-standard approach to determining the short-term efficacy and safety of therapies. In addition, agents with novel mechanisms of action for the treatment of chronic conditions, such as RA, require longterm data to better understand the risk-benefit profile over time.
Therefore, patients from qualifying phase I, phase II, and phase III index studies were eligible to participate in 1 of 2 open-label, longterm extension (LTE) studies: A3921024 (global) and A3921041 (Japan). Here we report pooled data from these LTE studies, describing the safety and tolerability of tofacitinib and the durability of the clinical response up to 48 months in patients with moderate to severe active RA.
MATERIALS AND METHODS
Index studies
The phase II population described in this analysis consisted of patients from 1 open-label and 8 double-blind, RCT index studies of 6 to 24 weeks’ duration. Tofacitinib was dosed between 1 and 30 mg BID as monotherapy (NCT00147498, NCT00550446, NCT00687193, NCT01059864, NCT01164579 [ongoing], NCT01359150)5,6,8,15,16 or in combination with background methotrexate (MTX; NCT00413660, NCT00603512, NCT01359150, NCT00976599)4,7,16,17. One of these studies (NCT00413660) had a 20 mg once daily (QD) arm in combination with background MTX, and another study (NCT00550446) had an active-control arm of adalimumab 40 mg subcutaneously every 2 weeks (q2w) as monotherapy. In addition, patients with RA enrolling into the LTE studies from a phase I open-label study of tofacitinib 10 mg BID (NCT01262118)18 were included. Patients from the phase II studies NCT00147498, NCT00550446, NCT00687193, NCT00413660, and NCT00603512 initiated treatment in the LTE studies with tofacitinib 5 mg BID. Patients from the other phase I and II studies initiated treatment in the LTE studies with tofacitinib 10 mg BID.
The phase III population consisted of patients from 6 double-blind, RCT (index studies) of 6 to 24 months’ duration. Tofacitinib was dosed at 5 or 10 mg BID as either monotherapy (NCT00814307, NCT01039688)11,12 or in combination with nonbiologic disease-modifying antirheumatic drugs (DMARD; mainly MTX; NCT00847613, NCT00960440, NCT00856544, NCT00853385)9,10,13,14 in patients with an inadequate response to 1 or more nonbiologic or biologic DMARD, including tumor necrosis factor (TNF) inhibitors. Patients randomized to placebo were advanced in a blinded fashion to tofacitinib 5 or 10 mg BID at 3 or 6 months. One phase III study (NCT0853385) had an active-control arm of adalimumab 40 mg subcutaneously q2w on background MTX. All patients from phase III index studies initiated treatment in the LTE studies with tofacitinib 10 mg BID regardless of treatment assignment in the index study, with the exception of patients from China and Japan who initiated treatment with tofacitinib 5 mg BID as required by the protocol.
Key patient inclusion criteria
Patients eligible for enrollment in the LTE studies were aged ≥ 18 years (A3921024) or ≥ 20 years (A3921041) with a diagnosis of RA based on the American College of Rheumatology (ACR) 1987 Revised Criteria, and had completed participation in a prior qualifying phase I, phase II, or phase III index study of tofacitinib for the treatment of RA.
Key exclusion criteria
Key exclusion criteria for the LTE studies included confirmed hemoglobin (Hb) < 9 g/dl or hematocrit < 30%; white blood cell count < 3.0 × 109/l; absolute neutrophil count < 1.2 × 109/l; platelet count < 100 × 109/l; estimated glomerular filtration rate < 40 ml/min (Cockcroft-Gault calculation); total bilirubin, alanine aminotransferase (ALT), or aspartate aminotransferase (AST) > 1.5 × upper limit of normal (ULN; A3921024) or > 2 × ULN (A3921041); treatment-related serious adverse events (SAE) in the index study; serious chronic or recurrent infections, including active or inadequately treated latent Mycobacterium tuberculosis, herpes zoster, or other opportunistic infection; evidence or history of malignancy, with the exception of adequately treated or excised non-metastatic basal or squamous cell cancer of the skin or cervical carcinoma in situ; or any lymphoproliferative disorder, history of lymphoma, leukemia, or signs and symptoms suggestive of current lymphatic disease.
Study design and treatment
The studies (A3921024 and A3921041) are registered with ClinicalTrials.gov (NCT00413699 and NCT00661661, respectively) and are conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines established by the International Conference on Harmonization. The final protocols, amendments, and informed consent documentation were reviewed and approved by the Institutional Review Boards and/or Independent Ethics Committee of each study center. All patients provided written informed consent.
Studies A3921024 (global) and A3921041 (Japan) are ongoing, multicenter, open-label LTE studies that include patients who have participated in a prior qualifying index study of tofacitinib. Both studies (A3921024 and A3921041) involve similar patient populations (other than race and its associated characteristics) and thus were deemed appropriate to be pooled.
Patients from the qualifying index studies initiated treatment in the LTE studies with oral tofacitinib 5 mg BID or 10 mg BID, as described above. For safety reasons, the tofacitinib dose could be reduced from 10 to 5 mg BID. For reasons of inadequate response, the tofacitinib dose could be increased from 5 to 10 mg BID. Tofacitinib dose adjustments were at the discretion of the investigator and could be temporary or last for the duration of the study. Dose adjustments to permitted concomitant RA medications [including MTX, leflunomide, sulfasalazine, antimalarials, auranofin, injectable gold preparations, nonsteroidal antiinflammatory drugs (NSAID) and/or glucocorticoids at approved doses] were at the discretion of the investigator for inadequate efficacy or safety reasons, or could be tapered or discontinued in response to adequate RA disease control.
Baseline values in the current analysis were those of the qualifying index study for patients who enrolled in the LTE studies within 14 days of the final visit of the index study; for other patients, baseline values were derived from the final predrug visit (e.g., LTE baseline visit) on entry to the LTE studies. For the current analysis, patients were classified as 5 or 10 mg BID based on the highest dose administered during the first 135 days of treatment in the LTE studies, and as background DMARD or monotherapy based on the requirements of the index study. Other conventions such as total daily dose were explored, and found to be in very good agreement with the 135-day rule — about 95% of patients retained their dosing classification as for the 135-day rule. Monotherapy refers to tofacitinib administered in the absence of any other DMARD except antimalarials. Safety data included observations over 60 months; efficacy data were included up to Month 48. All data retrieved up to and including April 19, 2012, were included in this interim analysis. Data collection and analyses are ongoing; therefore, the study database has not yet been locked (i.e., some values may change for the final, locked study database).
Objectives/endpoints
The primary objectives were to determine the longterm safety and tolerability of tofacitinib dosed at 5 and 10 mg BID. The primary endpoint was safety, which included the assessment of the type, severity, and frequency of AE, and changes observed in laboratory safety tests, irrespective of whether they were reported as an AE. The secondary objective was to evaluate the persistence of efficacy of tofacitinib dosed at 5 and 10 mg BID. Key secondary endpoints included ACR20, ACR50, and ACR70 response rates, 28-joint Disease Activity Score (DAS28)-4[erythrocyte sedimentation rate (ESR)] assessments, including mean change from baseline and rates of DAS-defined remission (DAS28-4-ESR < 2.6) and low disease activity (LDA; defined as DAS28-4-ESR ≤ 3.2), and mean change from baseline in Health Assessment Questionnaire-Disability Index (HAQ-DI). Safety and efficacy data were summarized descriptively.
Statistical analysis
Exposure-estimated incidence rates (IR) and exposure-adjusted event rates (EAER) were calculated as the number of unique patients with an event (for that time period), divided by the total exposure in that treatment group in the pooled cohort, and multiplied by 100. Note that, for the analysis for each individual event, the IR censors patient exposure at the time of event; EAER does not.
All safety and efficacy data reported in the text are pooled data (i.e., tofacitinib 5 and 10 mg BID ± background DMARD). Data for individual groups (dose; treatment regimen) can be found in the tables and figures.
RESULTS
Patients
In total, 4102 patients were treated in the LTE studies for a total of 5963 patient-years over a mean (maximum) duration of 531 (1844) days, which does not include initial exposure in the index study (Table 1). The numbers of patients treated with tofacitinib 5 mg BID and 10 mg BID per year were 284 and 1219 (Year 1), 345 and 1376 (Year 2), 194 and 83 (Year 3), 536 and 3 (Year 4) and 62 and none (Year 5), respectively. Patients in the 5 mg BID group (mainly from phase II index studies) had a longer treatment duration and greater exposure than patients in the 10 mg BID group (from the later phase III studies). Most patients (n = 2742; 66.8%) received background nonbiologic DMARD, while 33.2% of patients (n = 1360) received tofacitinib as monotherapy. Essentially all patients (n = 4043; 98.6%) received symptom-controlling treatments, such as NSAID and/or glucocorticoids; 2216 patients (54.0%) received glucocorticoids. The majority of patients (n = 3783; 92.2%) enrolled within 14 days of the final visit of the index study and therefore had baseline values from the index study, which defined the LTE baseline. For the remaining patients (n = 319; 7.8%), baseline values were collected before initiation of study treatment in the LTE studies or were incomplete if not executed per protocol. Patient baseline demographics are summarized in Table 1.
Patient demographics and baseline characteristics.
Safety
A safety summary is presented in Table 2. A total of 13,932 treatment-emergent AE were reported in 3152 patients (76.8%) with an IR of 154.5 events per 100 patient-years. Discontinuations occurred in 852 patients (20.8%).
Summary of safety data.
Discontinuations due to AE occurred in 437 patients (10.7%), with an IR of 7.3 events per 100 patient-years (Table 2). The most frequently reported classes of treatment-emergent AE leading to discontinuation by Medical Dictionary for Regulatory Activities (MedDRA) system organ class were infections and infestations, investigations (e.g., blood creatinine increased; ALT increased; AST increased), and neoplasias benign, malignant, and unspecified [including cysts and polyps (Table 2)]. SAE were reported in 630 patients (15.4%), with an IR of 11.1 events per 100 patient-years. Thirty-one deaths occurred overall, with an IR of 0.5 (95% CI: 0.36–0.73) events per 100 patient-years. Overall, rates of AE did not vary considerably over time.
The most frequently reported classes of treatment-emergent AE by system organ class (SOC) were infections and infestations (n = 2083; 50.8%), gastrointestinal (GI) disorders (n = 968; 23.6%), and musculoskeletal and connective tissue disorders (n = 960; 23.4%).
In the infection and infestation SOC, the 3 most frequent AE by MedDRA-preferred term were nasopharyngitis, upper respiratory tract infection, and urinary tract infection (Table 2). IR per 100 patient-years were calculated for infections of special interest, including opportunistic infections, tuberculosis, and herpes zoster (Table 2). Asian patients exposed to tofacitinib had a higher IR of herpes zoster infection compared with white patients (6.7 events per 100 patient-years vs 3.5 events per 100 patient-years, respectively).
Cardiovascular (CV) events were blindly adjudicated by an independent Cardiovascular Safety Endpoint Adjudication Committee. IR per 100 patient-years for CV AE of special interest ranged from 0.05-0.3 (Table 2).
IR per 100 patient-years for all malignancies (excluding nonmelanoma skin cancer) are summarized in Table 2, and were 0.5 (95% CI: 0.38–0.75) for nonmelanoma skin cancers.
In the GI SOC, the most common AE by MedDRA-preferred term were diarrhea (n = 179; 4.4%), nausea (n = 128; 3.1%), and gastritis (n = 101; 2.5%). Potential cases of GI perforation were reviewed across the entire tofacitinib RA development program (including phase II and phase III index studies), then adjudicated by 2 sponsor-employed gastroenterologists who independently evaluated and analyzed each case. Each case was classified as definite GI perforation (compatible AE term with sufficient supportive clinical evidence), probable GI perforation (compatible AE term but without sufficient supportive clinical evidence), or no GI perforation (no compatible AE term and no supportive clinical evidence; alternative diagnosis supported). Eleven cases of probable or definite perforation occurred in tofacitinib-treated patients; 1 case in the 3 mg BID group in a phase II study, 2 cases in the 10 mg BID group in phase III studies, and 8 cases in the LTE studies (5 mg BID: n = 4; 10 mg BID: n = 4; Table 3). Ten of 11 cases occurred in patients who received background MTX. Ten cases occurred in patients receiving background steroids. Six of the 11 perforations were reported as diverticular, and were most likely associated with diverticulitis upon investigation at the time of the perforation. The IR for GI perforation in the tofacitinib RA program was 0.13 (95% CI: 0.07, 0.24) events per 100 patient-years.
Definite or probable cases of gastrointestinal perforations in the tofacitinib program.
Changes in laboratory variables from baseline were observed, including changes in Hb, creatine kinase, AST, and ALT; decreases in neutrophil, lymphocyte, and platelet counts; increases in low-density and high-density lipoprotein cholesterol; and changes in serum creatinine (Figure 1A–E). Mean overall values for laboratory safety variables generally stabilized over time, with longer treatment duration in all tofacitinib groups. With the exception of an absolute lymphocyte count < 500 cells/mm3, which has been associated with an increased risk of serious infections, there was no clear relationship between changes in laboratory variables and clinical safety outcomes.
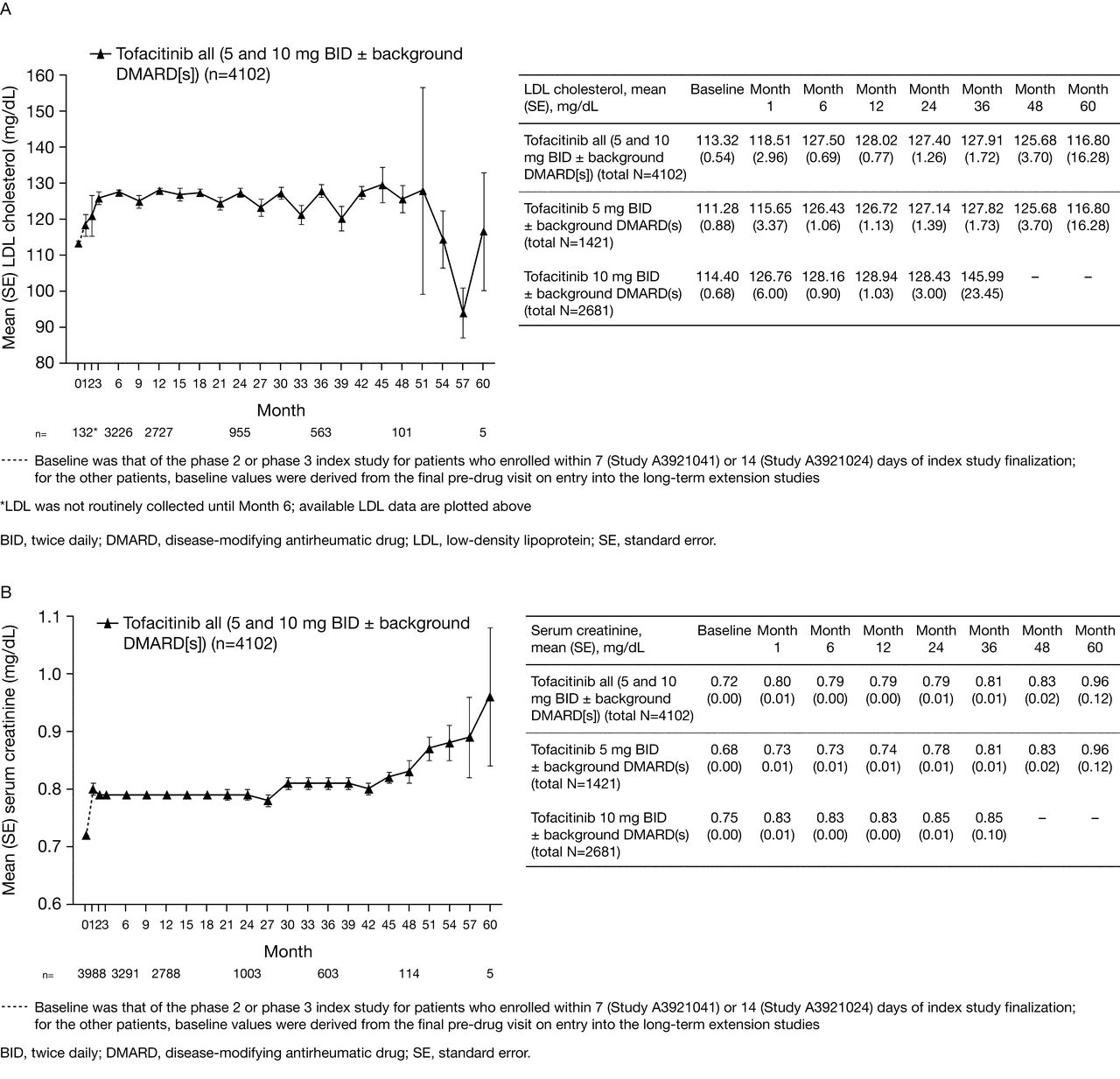


Mean (A) LDL cholesterol, (B) serum creatinine, (C) hemoglobin, (D) neutrophil counts, (E) lymphocyte counts, and (F) proportion of patients with mild neutropenia over time.
In the LTE studies, a total of 41 out of 4095 patients (1.0%) had a confirmed (2 consecutive measurements) decrease in Hb from baseline that met potentially life-threatening criteria. Of these 41 patients, 3 had decreased Hb (below lower limit of normal based on Covance central laboratory reference ranges) at baseline. Onset of decreased Hb levels ranged from 29 to 1184 days following start of tofacitinib treatment. The duration of decreased Hb levels ranged from 2 to 408 days. In the majority (26 patients), the decreased Hb either improved or returned to baseline levels, with the discontinuation of study drug (7 patients), or while remaining on study drug (19 patients). In 10 of 11 patients with sustained decreases, the Hb levels had stabilized; 1 patient experienced a progressive decrease in the setting of septic arthritis and shock, which resulted in a fatal outcome. Followup Hb levels were missing in 4 patients as of the cutoff date for entering data.
Fewer than 5% of tofacitinib-treated patients experienced neutropenia of any degree at any visit throughout the study, and most cases were mild in severity (Figure 1F). There were 30 out of 4095 patients (0.7%) with a confirmed (2 consecutive measurements) moderate to severe decrease in neutrophil cell count (< 1.5 × 103 cells/mm3 but ≥ 0.5 × 103 cells/mm3) in the LTE studies; no patients had a confirmed neutrophil count < 0.5 × 103 cells/mm3. Of these 30 patients, 6 had a decreased neutrophil count (below lower limit of normal of 1.96 × 103 cells/mm3 based on Covance central laboratory) at baseline. Onset of neutropenia ranged from 11 to 1268 days following start of tofacitinib treatment. The duration of neutropenia ranged from 6 to 342 days. In the majority of cases (n = 21), the decreased neutrophil counts either improved or returned to baseline counts. In 5 patients, the decreased neutrophil counts improved or returned to baseline counts with either temporary or permanent discontinuation of study drug. In 16 patients, the decreased neutrophil counts appeared to improve spontaneously without changing the dose of study drug. Of the 9 patients with a sustained decrease, only 1 patient, who had an abnormal baseline neutrophil count of 0.69 × 103 cells/mm3, experienced a sustained decreased neutrophil level of < 1.0 × 103 cells/mm3.
In total, 48 of 4053 patients (1.2%) had confirmed (2 consecutive measurements) increased ALT levels > 3 times the ULN in the LTE studies. Of 46 patients with available baseline data, 13 had increased ALT levels (above the ULN based on Covance central laboratory reference range) at baseline. Onset of increased ALT levels ranged from 14 to 1361 days following start of tofacitinib treatment. The duration of increased ALT levels was from 3 to 391 days. In most patients (n = 36), the increased ALT levels improved or returned to baseline levels. In 22 patients, the ALT levels improved or returned to baseline levels following discontinuation of study drug as per protocol requirements or at the discretion of the investigators. In 14 patients, the ALT levels appeared to improve spontaneously without discontinuing the study drug. Twelve patients had a sustained increase in ALT. For 3 patients with a sustained increase, the last reported ALT value was > 3 × ULN as well as the highest value measured. Of these 3 patients, 1 patient was diagnosed with chronic cholecystitis, which resolved after cholecystectomy (study drug was permanently discontinued prior to the surgery), 1 patient did not have an AE reported by the investigator and had not discontinued the study as of the cutoff date for data, and 1 patient experienced possible autoimmune hepatitis, although drug-induced liver injury (DILI) could not be ruled out (see below).
One patient receiving 10 mg BID tofacitinib and MTX experienced asymptomatic aminotransferase elevations on study, and both drugs were discontinued, without normalization of the aminotransferases. Two to 3 months later the patient developed jaundice in association with further increases in aminotransferase levels. The elevated liver tests responded to prednisolone and azathioprine, possibly consistent with autoimmune hepatitis, but DILI could not be ruled out.
A summary of changes in selected laboratory variables is presented in Table 2.
Efficacy
Efficacy over time was calculated using baseline values from the index (92.2%) or LTE studies (7.8%), as described in Materials and Methods.
ACR20, ACR50, and ACR70 response rates were sustained over time between Months 1 and 48 (Figures 2A–C). At Month 1, ACR20, ACR50, and ACR70 response rates for patients were 72.5% (n = 2923/4032), 48.0% (n = 1937/4032), and 27.4% (n = 1104/4032), respectively; corresponding rates at Month 48 were 74.4% (n = 96/129), 49.6% (n = 64/129), and 34.1% (n = 44/129).
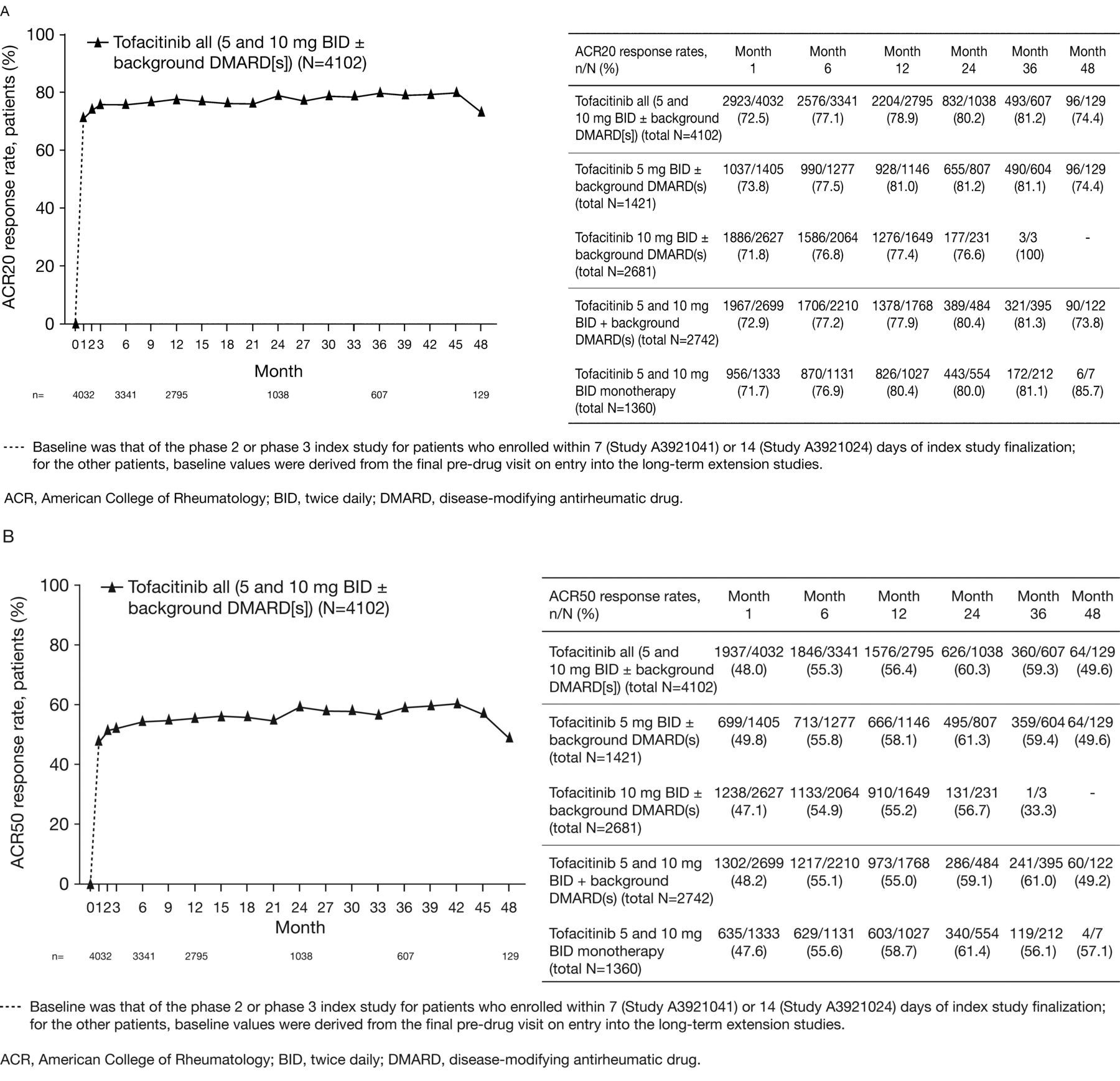

(A) American College of Rheumatology response rate: ACR20, (B) ACR50, and (C) ACR70 response rates over time.
Mean DAS28-4-ESR decreased from 6.3 at baseline to approximately 3.6 at Month 48 (Figure 3A). Rates of DAS-defined remission and LDA over time are shown in Figures 3B and 3C.
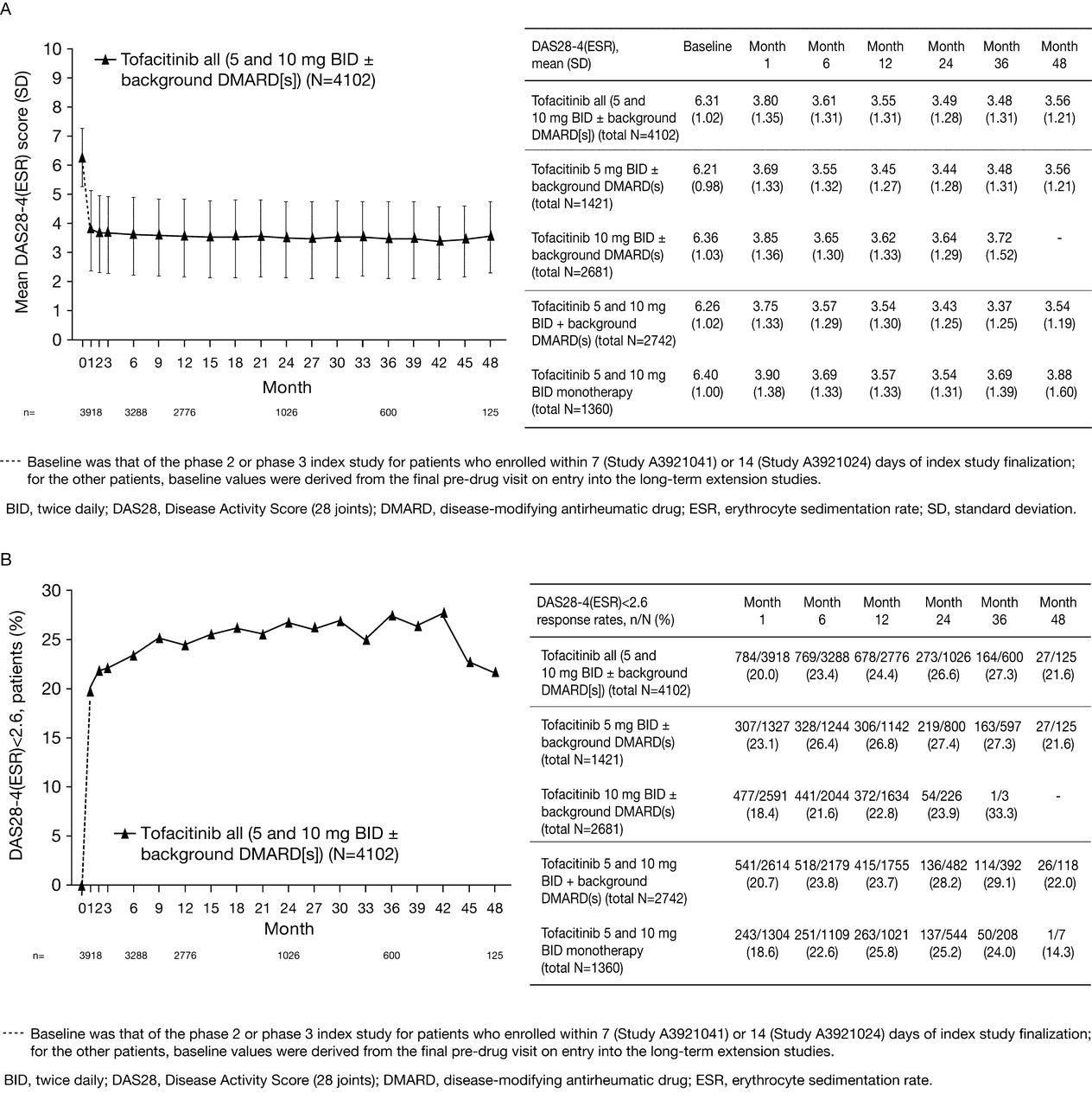

(A) Mean 28-joint Disease Activity Score (DAS28)-4[erythrocyte sedimentation rate (ESR)], (B) DAS-defined remission (DAS28-4-ESR < 2.6), (C) low disease activity (LDA; DAS28-4-ESR ≤ 3.2), and (D) mean Health Assessment Questionnaire Disability Index (HAQ-DI) over time.
Mean HAQ-DI score improved from 1.4 at baseline to 0.8 at Month 48 (Figure 3D).
DISCUSSION
The 2 LTE studies described here are part of one of the largest clinical development programs undertaken in the investigation of a drug for the treatment of RA to date. The current analysis presents the safety and efficacy of up to 4 years of exposure to open-label tofacitinib therapy administered as monotherapy (one-third of patients) or with nonbiologic DMARD in patients with moderate to severe active RA following participation in a qualifying phase I, phase II, or phase III index study.
Although the data reported here included patients experiencing up to 60 months of exposure to tofacitinib in the LTE studies, the overall 10 mg BID data after 24 months and monotherapy data after Month 36 are limited, and interpretations should therefore be made with caution. When pooling all patients regardless of dose group and treatment regimen, the number of patients is limited after Month 48 and consequently efficacy data are not reported beyond this timepoint. For the completeness of safety reporting, safety data are included up to 60 months of observation. Further, for the current analysis patients were classified as tofacitinib 5 or 10 mg BID based on the highest dose administered during the first 135 days of treatment in the LTE studies. This methodology does not account for cumulative exposure to treatment or any dose changes over time on therapy, although the number of patients who adjusted dose for > 14 days was small. Exposure in the index study was also not included.
The safety profile of tofacitinib in the LTE studies was generally consistent with that observed in previous phase II4,5,6,7,8 and phase III studies9,10,11,12,13,14, and no new safety signals were detected. With the exception of serious infections, where the rate was higher in the 10 mg BID dose group compared with the 5 mg BID dose group, no dose dependencies were observed for safety events of special interest in the LTE portion of the clinical development program. Importantly, the duration of treatment in the LTE studies was longer for patients receiving tofacitinib 5 mg BID compared with 10 mg BID, resulting in greater overall exposure to the 5 mg BID dose in the LTE studies. This was because patients from the earlier phase II index studies, who had the longest duration of exposure at the time of the data cutoff, initiated treatment with 5 mg BID, whereas patients from the later phase III index studies (Chinese and Japanese patients excluded) initiated treatment with 10 mg BID in the LTE studies. A dose-dependency for serious infections was not observed in phase III studies where the incidence was about 3 events per 100 patient-years for both doses19. The safety profile of patients receiving tofacitinib as monotherapy or with background DMARD was similar except for the incidence of liver enzyme abnormalities, for which combination treatment with background DMARD was associated with a higher rate. However, comparisons of data between monotherapy and combination treatment regimens should be treated with caution because each index study defined the regimen for all patients within that study and patients were not randomized to 1 regimen versus the other within any study.
Rates for safety events of special interest generally remained stable between phase III and LTE studies and were consistent with the published rates for TNF inhibitors and other biologic DMARD in similar RA populations for mortality20,21,22, serious infections including opportunistic infections and tuberculosis23,24,25,26,27,28,29,30,31,32,33,34,35,36, CV events37, and malignancies38. Patients with RA are at greater risk than the general population for a number of comorbidities, including CV events and malignancies. The relative risk inferred by the disease itself versus therapeutic interventions is not clearly understood. Enhanced pharmacovigilance for these events is therefore warranted. The IR for herpes zoster infection (serious and non-serious) was higher for tofacitinib [4.3 (95% CI: 3.83–4.90) per 100 patient-years] than the rates reported in the literature for patients with RA treated with biologic and nonbiologic DMARD (0.0034 to 2.4 events per 100 patient-years)39,40,41,42,43,44,45,46. It is noteworthy that the rates of herpes zoster in the phase III cohorts of patients receiving placebo and adalimumab in the tofacitinib program were also higher than those in the literature overall47,48. Recently reported risk factor analyses have shown that Asian patients exposed to tofacitinib had a higher IR of herpes zoster infection49. The rate of herpes zoster infection did not appear to increase after longterm treatment and followup48.
The IR for GI perforation in patients treated with tofacitinib [0.13 (95% CI: 0.07–0.24) events per 100 patient-yrs] was consistent with the previously published background rates for patients receiving biologic and nonbiologic DMARD (0.05 to 0.17 events per 100 patient-yrs50,51). The safety reported here relates only to findings in the LTE phase of the tofacitinib development program, with the exception of GI perforations, where an integrated analysis is presented (the majority of cases occurred in LTE studies). A detailed discussion of safety events of special interest is, however, warranted and will be addressed in dedicated safety reports in preparation, which will integrate safety findings from LTE, phase II, and phase III studies, to provide an integrated summary from across the development program37,38,48,49,52,53.
Changes in laboratory variables were generally consistent with observations in completed phase II and phase III studies. Mean overall values for laboratory safety variables generally stabilized over time, with longer treatment duration in all tofacitinib groups. Most cases of anemia were mild to moderate and the incidence of potential life-threatening anemia according to Outcome Measures in Rheumatology (OMERACT) criteria was small (1%) in the LTE studies. In the majority of patients with confirmed potentially life-threatening decreases in Hb, moderate to severe neutropenia, and increases in ALT > 3 × ULN, levels improved or returned to baseline either spontaneously or with discontinuation of the study drug.
Tofacitinib had a reversible effect on mean serum creatinine levels in patients with RA, and these changes plateaued, remained within normal limits (ULN: 1.1 and 1.3 mg/dl for females and males, respectively), and did not appear to be associated with acute renal failure or progressive worsening of renal function in the LTE studies. The assessment of laboratory variables is ongoing in the LTE studies.
A posthoc analysis was performed of safety findings between patients with an inadequate response to nonbiologic DMARD versus patients with an inadequate response to biologic DMARD54. Event rates for important safety events were generally similar across nonbiologic DMARD-IR and biologic DMARD-IR populations with small numerical differences noted in serious infections, and SAE favoring the nonbiologic DMARD-IR population. These numerical differences may be attributable to differences in baseline characteristics (e.g., age, race) between the nonbiologic DMARD-IR and biologic DMARD-IR populations. CI were wide and overlapping. Rates of discontinuations due to AE, herpes zoster, major adverse CV events, malignancies, and deaths were similar across populations54.
Concerning efficacy endpoints, tofacitinib administered as monotherapy or in combination with background DMARD provided sustained improvements over 48 months of longterm open-label treatment. The efficacy achieved in phase II and phase III studies was persistent as reflected in clinically meaningful reductions in the signs and symptoms of RA as measured by ACR20/50/70 response rates and DAS28-4-ESR, and improvements in physical function, as measured by HAQ-DI. However, changes in efficacy measures over time may have been affected by progressively smaller patient numbers and any effects of discontinuation.
It is recognized that LTE studies enroll only those patients who were eligible and completed the preceding randomized clinical trials, a patient population in whom the agent is known to be efficacious and well tolerated. Consequently, there are limitations to fully characterizing an agent’s benefit/risk profile in the setting of a clinical development program. This holds especially true for understanding the safety profile, in particular over the longer term and in more real-life conditions. Open-label LTE studies, such as those reported here, may nevertheless serve as important surrogates in the absence of longer term real-life data and play an important role in further describing the safety profile of drugs with novel mechanisms of action, such as tofacitinib.
Tofacitinib dosed at 5 or 10 mg BID in patients with RA demonstrated a consistent safety profile and sustained efficacy up to 48 months in open-label LTE studies, supporting a favorable risk-benefit profile for both doses. The safety and efficacy profile of tofacitinib monotherapy was consistent with that observed when tofacitinib was administered with background nonbiologic DMARD. These LTE studies are ongoing and their results will continue to be updated and reported.
Acknowledgment
The authors thank the patients who were involved in this study, the investigators, and the entire study team. Editorial support was provided by Karen Irving.
Footnotes
-
Full Release Article. For details see Reprints/Permissions at jrheum.org
-
Funded by Pfizer Inc. J. Wollenhaupt has served as a consultant and member of the Speaker’s bureau for Abbott, Chugai, Pfizer Inc., Roche, and UCB. J. Silverfield has received research support from Pfizer Inc. E.B. Lee has served as a consultant for Pfizer Inc. J.R. Curtis has served as a consultant for, and has received research support from Pfizer Inc.; S.P. Wood, K. Soma, C.I. Nduaka, B. Benda, D. Gruben, H. Nakamura, Y. Komuro, S.H. Zwillich, L. Wang, and R.J. Riese are employees of Pfizer Inc. and hold stock/stock options in Pfizer Inc. Editorial support was provided by Karen Irving at Complete Medical Communications and was funded by Pfizer Inc.
- Accepted for publication November 19, 2013.
Free online via JRheum Full Release option








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}