

Abstract
Objective. High mobility group box chromosomal protein 1 (HMGB1) has been implicated as a mediator of inflammation in rheumatoid arthritis (RA), while its role in juvenile idiopathic arthritis (JIA) has not been described. To evaluate the role of HMGB1 in the inflammatory process in JIA and its potential as a therapeutic target, we investigated whether extracellular HMGB1 is detectable in JIA and if so, to correlate the levels with established inflammatory markers and clinical measures.
Methods. Matching samples of blood and synovial fluid (SF) were collected from 23 patients with JIA. Levels of HMGB1, soluble receptor for advanced glycation endproducts, S100A12, myeloid-related protein 8/14, and other inflammatory mediators were analyzed.
Results. Significantly increased HMGB1 levels were recorded in SF compared to blood samples from patients with JIA. The amount of HMGB1 was highest in patients with early disease onset irrespective of disease duration. In contrast, the proinflammatory S100 protein and interleukin 8 were highest in patients in early phases of disease. Matrix metalloproteinase-3, a marker of cartilage destruction, was higher in patients with late disease onset, indicating similarities with RA in that patient subgroup.
Conclusion. Levels of extracellular HMGB1 are increased in the inflamed joints of patients with JIA. This warrants further studies of HMGB1 as a mediator of JIA pathogenesis as well as a biomarker for inflammatory activity and as a target for therapy. The variation in levels of HMGB1 and S100 proteins in relation to disease onset indicates a difference in inflammatory phenotype during disease progression.
Juvenile idiopathic arthritis (JIA) is a collective diagnosis for a number of chronic inflammatory disorders with diverse articular and extraarticular manifestations. JIA can be divided into 7 subgroups according to criteria from the International League of Associations for Rheumatology (ILAR). Common for all subgroups is onset before 16 years of age and persistent arthritis for at least 6 weeks1,2. The course of the disease varies between subgroups and individuals and, unlike adults with rheumatoid arthritis (RA), children with JIA have a significant chance to recover with minimal or no residual consequences of the disease3. Thus, a challenge for the pediatrician is to detect and treat overt inflammation and tissue destruction while avoiding potential side effects of the medications in the growing child.
Knowledge of the mechanisms involved in progression of JIA and the immunological differences between the subgroups remains incomplete. Hence, to develop new therapies and to define reliable biomarkers of inflammation and joint destruction that would enable more precise monitoring of disease progression, there is a need to better identify the inflammatory phenotype of patients with JIA.
The majority of children with JIA respond well to conventional treatment or treatment with biologics, e.g., tumor necrosis factor inhibitor (anti-TNF) and anti-interleukin 1 (IL-1) therapy, but none of the drugs currently used has a completely curative potential. Thus, further understanding of the inflammatory processes in JIA is necessary for the definition of new potential therapeutic targets.
High mobility group box chromosomal protein 1 (HMGB1) is a DNA-binding protein present in all nucleated cells. During cellular stress and cell death, HMGB1 can be released into the extracellular space. Similarly, activated myeloid cells secrete HMGB1. Once extracellular, HMGB1 acts as an endogenous danger signal, promoting inflammation. By interaction with Toll-like receptors TLR4, TLR2, and the receptor for advanced glycation endproducts (RAGE), HMGB1 mediates either cytokine production or cell migration4,5. The receptor interactions and the extracellular function of HMGB1 in different stages of inflammation are regulated by the redox status of HMGB1’s 3 redox-sensitive cysteines. The HMGB1 released during tissue injury mainly promotes cell recruitment, whereas the HMGB1 actively released during the inflammatory phase induces cytokine production. During the resolution of inflammation, HMGB1 is oxidized and does not induce either cell migration or cytokine production4,6,7,8.
In addition to its direct inflammation-promoting functions, HMGB1 has the potential to form complexes with certain molecules and thereby to strongly enhance the inflammatory response to these ligands. Of particular interest from a rheumatological viewpoint is the interaction of HMGB1 with IL-1α, IL-1β, and nucleosomes. We have previously shown that HMGB1 in complex with IL-1α and IL-1β significantly enhanced the production of proinflammatory mediators and matrix metalloproteinases in synovial fibroblasts from patients with RA9.
Three fundamental findings indicate that extracellular HMGB1 plays an important role in arthritis. First, HMGB1 is aberrantly expressed in the synovial tissue of patients with RA and increased levels are recorded in synovial fluid (SF)10. Second, intraarticular injections of recombinant HMGB1 result in development of arthritis in healthy mice11. Third, therapeutic HMGB1-specific blockade suppresses disease development in several experimental arthritis models10,12,13,14.
To date, the role of HMGB1 in JIA has not been studied and there are no reports, to our knowledge, describing the presence of extracellular HMGB1 in SF, plasma, or sera from patients with JIA. As a first step, we aimed to define whether increased levels of extracellular HMGB1 can be detected in patients with active JIA, and if so, to assess the relationship between HMGB1 levels and inflammatory mediators as well as clinical disease measures. Our study forms a basis for further evaluation of HMGB1, its different isoforms, and its role as a biomarker and a potential therapeutic target in patients with JIA. Additionally, our study reports a biomarker profile consisting of several inflammatory mediators that can be used to characterize different disease stages.
MATERIALS AND METHODS
Patients with JIA and healthy controls
Included in the study were 23 patients with JIA, median age 12 years (range 2–18), with median disease duration 10 months (range 0–180), and age at diagnosis 6 years (range 1–16). All had active disease with synovial effusion and a need for corticosteroid injections. SF and parallel plasma samples were collected from 15 patients at Astrid Lindgren Children’s Hospital, Stockholm, Sweden. SF and parallel serum samples were collected from 8 patients at Tartu University Hospital, Children’s Clinic, Tartu, Estonia. Patients had oligoarticular, polyarticular, enthesitis-related, or other type of JIA according to the ILAR criteria1. Clinical data for all patients are summarized in Table 1. Serum samples were collected from 10 healthy controls (7 males and 3 females), age 8 years (range 2–14), at Astrid Lindgren Children’s Hospital. The study was approved by the local ethics committees in Sweden (Stockholm) and Estonia (University of Tartu). Parents and patients gave informed consent. Data obtained from plasma and sera samples have been pooled because no significant differences of HMGB1 (p = 0.1627), soluble RAGE (sRAGE; p = 0.2766), and matrix metalloproteinase-3 (MMP-3; p = 0.9616) levels could be detected upon comparison (data available from the authors on request), and henceforth they are referred to as plasma.
Clinical data for all patients.
Separation of serum, plasma, and SF.
Plasma was obtained from blood samples collected in EDTA tubes and sera from blood samples collected in tubes without additive. SF samples were collected in citrate tubes or tubes without additive. Blood samples and SF were centrifuged at 1440 g for 10 min within 4 h and cells were removed. Sera/plasma/cell-free SF samples were stored at −80°C until assayed.
ELISA for detection of HMGB1, MMP-3, sRAGE, and cartilage oligomeric matrix protein (COMP)
All ELISA were performed according to the manufacturer’s instructions. HMGB1 levels were measured in serum/plasma (undiluted) and SF (diluted 1:10) from patients with JIA using HMGB1 ELISA kit II (IBL International). The lower limit of detection was 0.3 ng/ml.
MMP-3 levels were measured in serum/plasma (diluted 1:10) and SF (diluted 1:10,000 and 1:100,000) from patients with JIA using human MMP-3 ELISA DuoSet kits (R&D Systems). The lower limit of detection was 310 pg/ml for serum/plasma and 310 ng/ml for SF.
Levels of sRAGE were measured in serum/plasma (diluted 1:20) and SF (diluted 1:2 and 1:20) from patients with JIA using human RAGE ELISA DuoSet kits or human RAGE Quantikine ELISA Kit (R&D Systems). The lower limit of detection was 125 pg/ml.
COMP levels in SF (diluted 1:1000) from patients with JIA were measured by Cartilage Oligomeric Matrix Protein Human ELISA (Biovendor).
ELISA for detection of myeloid-related protein (MRP) 8/14 and S100A12
MRP8/14 and S100A12 levels in SF from patients with JIA were measured by an in-house sandwich ELISA, as reported15.
Cytometric bead array for detection of cytokines and chemokines
Levels of IL-12p70, TNF, IL-10, IL-6, IL-1β, and IL-8 were measured in SF (diluted 1:5 and 1:25) from patients with JIA by cytometric bead arrays using the Human Inflammatory Cytokines Kit (B&D Biosciences). Levels of monocyte chemotactic protein-1 (MCP-1), interferon gamma-induced protein (IP-10), regulated on activation normal T cell expressed and secreted (RANTES), interferon-γ (IFN-γ), IFN-α, and IL-17A were measured in SF (diluted 1:2 to 1:32) by cytometric bead arrays using Human Soluble Protein Flex Sets and Human Soluble Protein Master Buffer Kits (B&D Biosciences). All analyses were performed according to the manufacturer’s instructions. The detection limit for the selected cytokines was 100 pg/ml for the Inflammatory Cytokines Kit and 20 pg/ml for Flex Sets.
Detection of lactate dehydrogenase (LDH)
Levels of LDH were analyzed at the clinical laboratory at Karolinska University Hospital.
Statistical analysis
Data were analyzed using GraphPad Prism (GraphPad Software Inc.). Wilcoxon signed-rank test for paired samples was used to analyze the differences between SF samples and paired plasma samples, taken at the same timepoint. The relationships between levels of HMGB1 and investigated analytes and with clinical measurements were analyzed by Spearman’s rank correlation test. The values are expressed as medians and interquartile range (IQR). P value < 0.05 was considered statistically significant.
RESULTS
HMGB1 levels in patients with JIA and controls
To define a potential role for HMGB1 in JIA pathogenesis we determined the levels of secreted HMGB1 in SF and plasma samples from 23 patients with JIA and in serum from 10 healthy children.
There was no significant difference between HMGB1 levels in plasma/serum samples from patients with JIA and those from controls (Figure 1A). In contrast, HMGB1 levels were significantly increased in the SF samples compared to the corresponding plasma samples; median levels were 57.9 (IQR 19.1–104) ng/ml versus 0.65 (IQR 0.11–1.43) ng/ml, respectively (Figure 1B). The plasma levels recorded in healthy children corresponded to the previously reported HMGB1 levels in plasma samples from healthy adult individuals16.
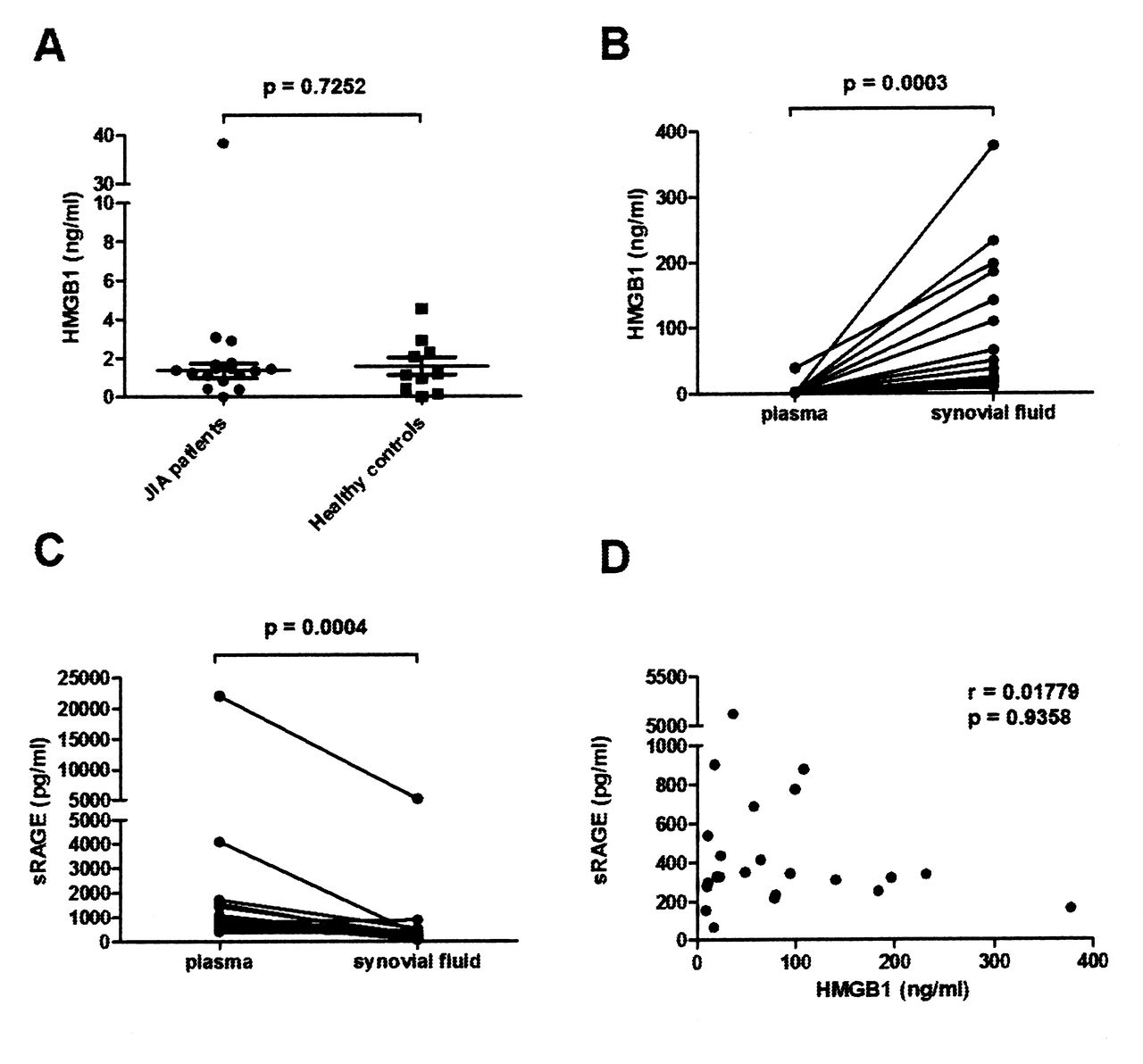
HMGB1 levels are increased and sRAGE levels decreased in synovial fluid (SF) compared to plasma. A. HMGB1 levels in plasma from patients with JIA and controls. B. HMGB1 levels in SF and plasma from patients with JIA. C. Levels of sRAGE in SF and plasma from patients with JIA. D. Correlation between levels of HMGB1 and levels of sRAGE in SF. Levels of HMGB1 and sRAGE were measured by ELISA and correlation was calculated using Spearman’s rank correlation test. HMGB1: high mobility group box chromosomal protein 1; sRAGE: soluble receptor for advanced glycation endproducts; JIA: juvenile idiopathic arthritis.
HMGB1 and sRAGE levels in SF from patients with JIA
One of the HMGB1 binding receptors, RAGE, exists as both a cell-bound and a soluble decoy receptor17. Decreased serum sRAGE levels in patients with JIA have been reported18 and it might be speculated that a regulatory mechanism for extracellular HMGB1 activity is compromised in patients with JIA. By defining sRAGE levels in both SF samples and plasma samples, we could confirm significantly lower sRAGE levels in SF compared to plasma. Median levels were 326 (IQR 265–486) pg/ml versus 810 (IQR 534–1434) pg/ml, respectively (Figure 1C). However, we did not reveal any inverse relationship of HMGB1 levels with sRAGE levels in the SF samples (Figure 1D).
Associations between S100 protein levels in JIA SF and IL-8, IL-6, and HMGB1
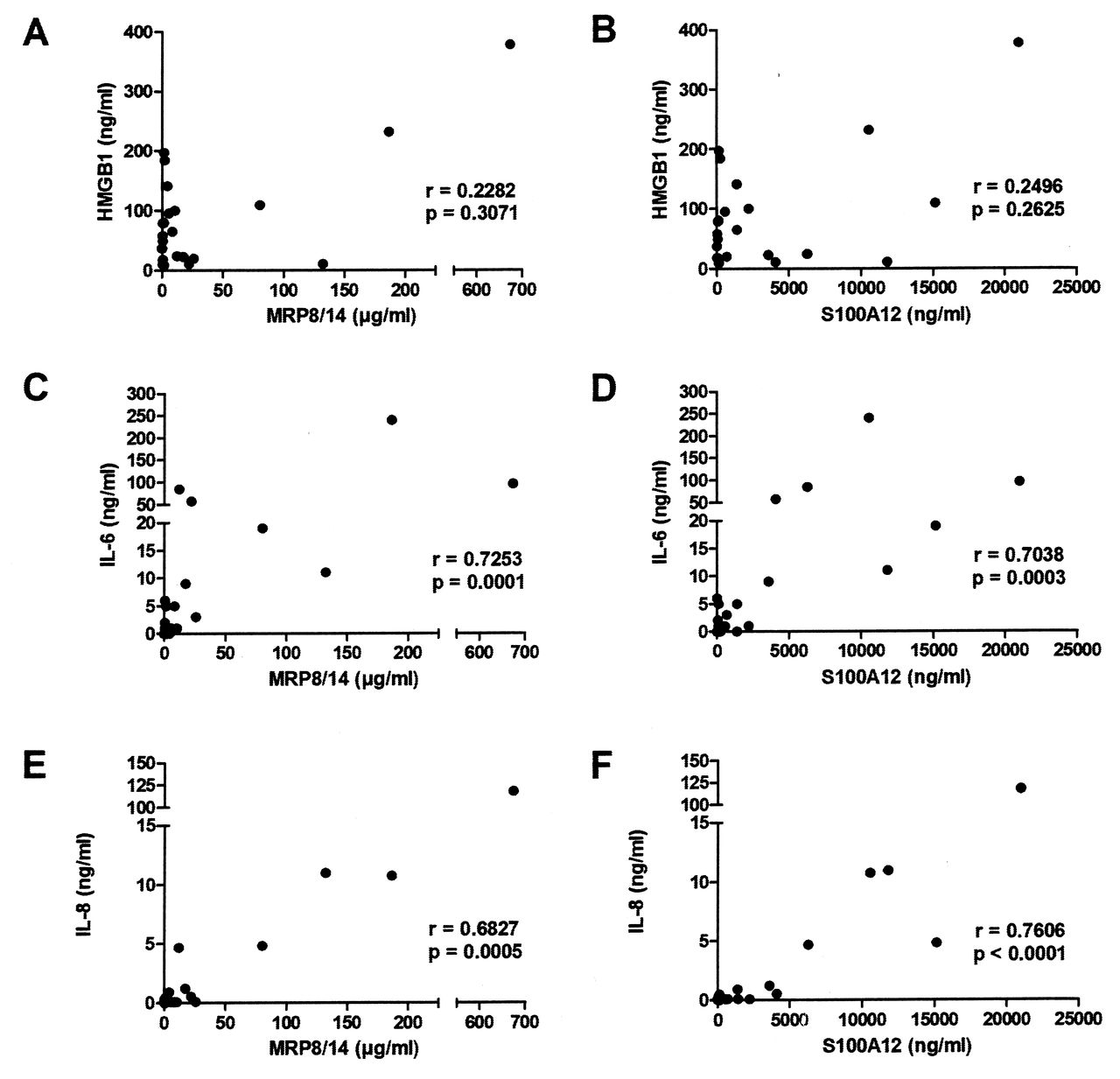
S100 proteins MRP8/14 and S100A12 have proinflammatory activity and their serum levels can be used as biomarkers for disease activity and treatment response in JIA19,20. Similarly to HMGB1, S100 proteins exist preformed inside the cells and can be released actively from neutrophils and myeloid cells as well as passively during cell death21. We detected high levels of S100 proteins in SF samples with median values of 5 μg/ml of MRP8/14 and 690 ng/ml of S100A12. However, there was a wide variation in levels between patients (Table 2). There was no correlation between the HMGB1 and S100 protein levels in SF samples, suggesting different induction/regulatory mechanisms for these 2 mediators (Figure 2A, 2B). Interestingly, the levels of both MRP8/14 and S100A12 correlated with IL-6 (Figure 2C, 2D), with IL-8 (Figure 2E, 2F), and with each other (data not shown).

Synovial fluid (SF) HMGB1 levels did not correlate to S100 proteins in contrast to IL-6 and IL-8. No correlation was seen between HMGB1 and (A) MRP8/14 and (B) S100A12 in SF. A significant correlation was observed between IL-6 levels and levels of (C) MRP8/14 and (D) S100A12 as well as correlation between levels of IL-8 and levels of (E) MRP8/14 and (F) S100A12 in SF. All correlations were calculated using Spearman’s rank correlation test. HMGB1: high mobility group box chromosomal protein 1; IL: interleukin; MRP: myeloid-related protein.
Levels of cytokines, chemokines, and inflammatory mediators in SF and plasma from patients with JIA were analyzed by ELISA or cytometric bead array.
Association between HMGB1 in JIA SF and cytokine and chemokine levels
We determined SF levels of mono-cyte- and T cell-derived cytokines and chemokines to evaluate whether there is a relationship between increased HMGB1 levels and inflammatory mediators produced locally in the joint. The concentration of cytokine/chemokine varied widely among individuals with IL-6 and IP-10, reaching the highest median values. In SF, we could not detect IL-12p70, TNF, IL-10, IFN-α, and IL-17A, and only 1 patient had detectable IFN-γ and IL-1β levels (Table 2). However, no statistically significant correlations could be determined between SF HMGB1 levels and IL-6, IL-8, IP-10, MCP-1, or RANTES, indicating a differential regulatory pathway for extracellular HMGB1 as compared to the investigated cytokines/chemokines (Figures 3A–3E).

HMGB1 levels in synovial fluid (SF) did not correlate to any measurements except for MMP-3. Correlation between levels of HMGB1 and levels of (A) IL-6, (B) IL-8, (C) IP-10, (D) MCP-1, (E) RANTES, (F) COMP, (G) LDH, and (H) MMP-3 in SF from patients with JIA as measured by cytometric bead arrays was calculated by Spearman’s rank correlation test. Panel I. MMP-3 levels in SF and plasma from patients with JIA as measured by ELISA. JIA: juvenile idiopathic arthritis; HMGB1: high mobility group box chromosomal protein 1; IL: interleukin; MMP: matrix metalloproteinases; MCP: monocyte chemotactic protein; IP: interferon gamma-induced protein; RANTES: regulated on activation normal T cell expressed and secreted; COMP: cartilage oligomeric matrix protein; LDH: lactate dehydrogenase.
Correlation between synovial HMGB1 concentrations and destruction markers
COMP, a noncollagenous extracellular matrix protein, is used as a marker of cartilage turnover22. To evaluate the relationship between HMGB1 and cartilage destruction in JIA, we measured the levels of COMP in SF from patients with JIA (Figure 3F). We found no significant correlation between synovial HMGB1 and COMP levels. This indicates that destroyed cartilage is not the main source of synovial HMGB1. LDH released from dying or injured cells is used as a marker for cell death and destruction. There was no association between HMGB1 and LDH levels in SF of patients with JIA (Figure 3G). This indicates that HMGB1 is not released exclusively from dying or injured cells.
MMP released by resident and infiltrating cells in the arthritic synovium are involved in tissue remodeling and contribute to the degradation of cartilage and bone. It has previously been demonstrated that MMP-3 levels are elevated in SF and sera in patients with JIA and that serum levels correlate with disease activity. MMP-3 is therefore suggested as a specific marker for disease activity23. We observed 1000-fold higher median levels of MMP-3 in the SF of patients with JIA as compared to corresponding plasma levels [17.0 (range 11.6–27.2) μg/ml vs 32.7 (range 20.4–38.7) ng/ml, respectively; Figure 3I] in agreement with previous studies23. Further, we revealed a significant negative correlation between HMGB1 levels and MMP-3 levels in SF (Figure 3H). Thus, HMGB1 might be involved in regulating mechanisms of destruction.
Correlation between inflammatory mediators and clinical disease measures
Next, to study whether the SF levels of HMGB1 and inflammatory biomarkers covaried with clinical disease measures, we analyzed the relationship between these markers with regard to disease duration, age at diagnosis, antinuclear antibody (ANA) positivity, and treatment. We could not observe any statistically significant association between HMGB1 levels and disease duration. In contrast to S100A12, MRP8/14, and IL-8 levels, which were higher in patients at disease onset, HMGB1 was present in SF in similar levels irrespective of disease duration (Figures 4A–4D). Again, this indicates a differential regulation of HMGB1 compared to the other biomarkers analyzed.
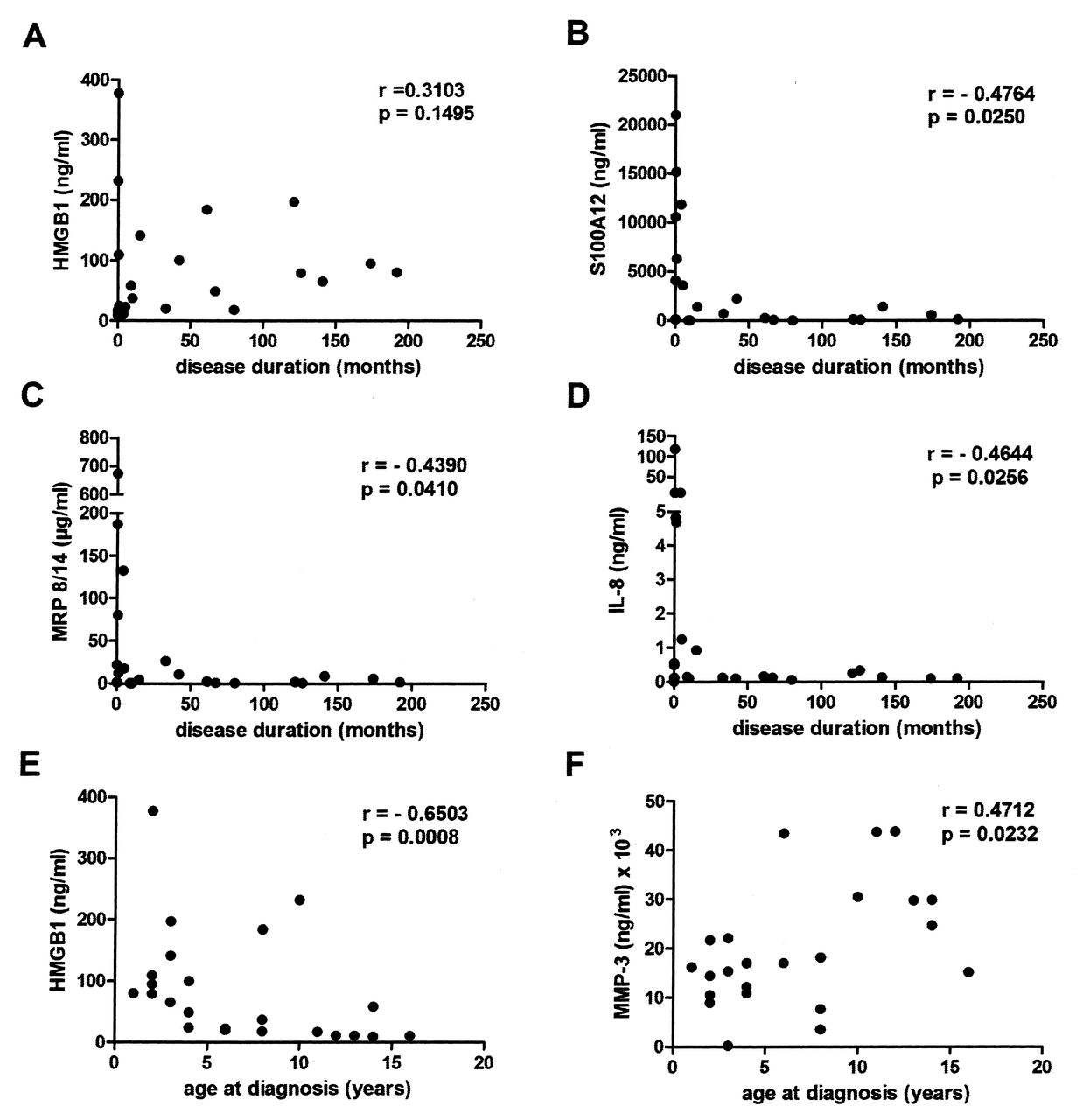
HMGB1 levels were higher in patients with early disease onset, independent of disease duration. Correlations were calculated by Spearman’s rank correlation test. Correlations between disease duration and levels of (A) HMGB1, (B) S100A12, (C) MRP8/14, and (D) IL-6 in synovial fluid (SF). Correlations between age at diagnosis and levels of (E) HMGB1 and (F) MMP-3 in SF. HMGB1: high mobility group box chromosomal protein 1; MRP: myeloid-related protein; IL: interleukin; MMP: matrix metalloproteinases.
Finally, we analyzed the associations between age at diagnosis of JIA and inflammatory biomarkers. We found a significant negative correlation between age at diagnosis and levels of HMGB1 in SF (Figure 4E), indicating that patients diagnosed with JIA at an early age had higher levels of HMGB1 throughout the active phases of the disease course than those with a later onset of JIA. Interestingly, the levels of tissue-degrading MMP-3 showed a positive correlation, with age at diagnosis being higher in those patients with a later disease onset (Figure 4F). There was no significant difference in levels of any of the analyzed mediators between ANA-positive and ANA-negative patients (data not shown), and the levels of analyzed mediators in the SF were not significantly influenced by any treatment (Table 1).
DISCUSSION
Multiple studies have implicated a pathogenic, proinflammatory role of extracellular HMGB1 in experimental arthritis models. Similarly, the reports of increased levels of HMGB1 in SF and in sera of patients with RA have evoked an interest in HMGB1 as a mediator of arthritis, as a biomarker for disease activity, and as a potential target molecule for therapy. To date, there are no reports exploring the involvement of HMGB1 in the pathogenesis of JIA. This is the first study examining the HMGB1 level and its relationship with inflammatory biomarkers in patients with JIA. We report that HMGB1 is present at high levels in SF as compared to plasma in patients with JIA. We also show that HMGB1 levels are higher in patients with disease onset at an early age, and that this is independent of disease duration. Further, we demonstrate that JIA patients with high levels of HMGB1 in SF have low levels of MMP-3, a proteinase involved in bone and cartilage degradation.
Considered with previous knowledge, our results support a pathogenic role of HMGB1 in JIA because the levels of HMGB1 are elevated within the inflamed joints. The levels recorded in JIA are in keeping with previous measurements in RA SF, and higher than those recorded in SF from patients with osteoarthritis24. The significant increase of HMGB1 locally in JIA SF as compared to plasma levels indicates that cells present in the inflamed joints are the main source of extracellular HMGB1. There were no correlations between HMGB1 levels and LDH, indicating that the major source of HMGB1 is not dying cells, but instead, a combination of passive and active release.
The presence of cytokines and chemokines in SF has been described in oligoarticular JIA and correlates well with the results recorded in our cohort25. Thus, despite the small size of our study group, it appears to be representative in this aspect. HMGB1 did not correlate with any of the chemokines and cytokines measured in SF. It is noteworthy that RANTES and MCP-1, which were present in high levels, can be produced by synovial fibroblasts in addition to production by leukocytes. It has also been demonstrated that synovial fibroblasts subjected to hypoxia release HMGB110, and tissue hypoxia is highly associated with the severity of experimental arthritis26. Local hypoxia has been demonstrated to influence JIA inflammation27,28. Taken together, this might indicate that hypoxic synovial cells, e.g., synovial fibroblasts, are strongly activated during JIA and are a possible major source of HMGB1 in the arthritic joint.
In a comparison of HMGB1 levels with S100 protein levels in SF, no significant correlation was found. The production of the investigated S100 proteins MRP8/14 and S100A12 is restricted to the monocytic and granulocytic cell lineages, and increased serum levels of S100A12 protein have been proposed as a biomarker to monitor JIA disease activity, in particular neutrophil activation19,20,21. Therefore, it is likely that the increased S100 protein levels recorded in our cohort reflect a monocytic/granulocytic and systemic inflammation. In contrast, the increased levels of HMGB1 might indicate the onsite cellular activation, including inflammatory activity of synovial fibroblasts. This is further corroborated by our findings that HMGB1 is locally increased to similar levels in the inflamed joint, irrespective of disease duration. In contrast, the highest levels of S100 proteins and IL-8 in SF were recorded in patients with short disease duration, whereas a significant positive correlation was also seen between these markers. IL-8 is a potent attractant for neutrophils, which are the most abundant cell type in the inflamed SF, and might thus contribute to the high release of S100 proteins early in the disease process. It has also been reported that HMGB1 interacts with phosphatidylserine on the surface of neutrophils and mediates inhibition of phagocytosis of apoptotic neutrophils29 and might thereby retard inflammation and contribute to release of inflammatory mediators from neutrophils.
Both S100 proteins and HMGB1 are passively released because of cell death in addition to their active secretion. Therefore, the contribution of ongoing cell death in the arthritic joints to the release of these mediators cannot be excluded. Regarding HMGB1, this could be studied by analyzing the acetylation status of extracellular HMGB1, with actively secreted HMGB1 being acetylated. The actively secreted acetylated form of HMGB1 can only be measured by analytical tandem mass spectrometry30, a time-consuming approach not practical for studying large patient cohorts and beyond the scope of our study.
Comparing HMGB1 with recorded levels of matrix-degrading MMP-3, we discovered that patients with high levels of HMGB1 had low levels of MMP-3 in SF. MMP-3 can be produced by synovial fibroblasts and is known to be upregulated by proinflammatory cytokines. MMP-3 has been suggested as a marker for destruction in RA31 and has also been described in JIA23,32. The levels of MMP-3 recorded in our study are in agreement with previous reports23,32. Further, production of MMP-3 was associated with older age at disease diagnosis. These results indicate similarities with late-onset JIA and RA, because higher levels of MMP-3 have been detected in SF from patients with RA31. Whether the negative correlation between the production of MMP-3 and the release of HMGB1 is due to differential regulatory mechanisms remains to be resolved.
Even though the recorded levels of HMGB1 in SF were independent of disease duration, HMGB1 was present at higher levels in patients with disease onset in their early years. This suggests a possibility that early-onset JIA may be a distinct subgroup with an etiopathogenesis different from that of late-onset JIA and from RA. This finding is supported by previous studies showing that JIA disease onset at a young age is one of the predictors for persistent disease and joint erosions33. Our data are supported by previous reports showing biologic differences between patients with early-onset and late-onset JIA, independent of classification based on the number of joints involved34. The reverse connection of HMGB1 and MMP-3 to age at diagnosis may explain the negative correlation between HMGB1 and MMP-3. The interrelationships of disease onset, HMGB1 levels, and MMP-3 levels deserve further study in larger cohorts with better-defined subgroups. Treatment did not appear to affect the amount of HMGB1 in SF; however, the small cohort size prevents any firm conclusions.
The findings of our study suggest that HMGB1 has a role in the pathogenesis of JIA because it is significantly increased in the inflamed joints of patients with JIA and thereby reflects the extent of local inflammation. We have also shown that HMGB1, in contrast to S100 proteins, does not correlate with inflammatory cytokines such as IL-6 and IL-8. These mediators may be more important at the beginning of the disease, while HMGB1 seems to be involved during active disease, independent of disease duration. These results suggest the need for a multi-biomarker approach because some markers are more closely related to disease activity and others are present all the time and can explain the chronicity of the disease.
The questions for future studies are (1) Which molecular forms of HMGB1 are present in the SF in JIA? and (2) What is their function in the inflammatory process? Thus, our study justifies more extensive research in patients with JIA regarding the role of HMGB1 as a biomarker for disease activity, progression, and destruction.
Acknowledgment
We thank the staff of the pediatric rheumatology clinic at Astrid Lindgren Children’s Hospital, Stockholm, Sweden, and the Children’s Clinic at Tartu University Hospital, Tartu, Estonia, for collection of samples. We also thank Heidi Wähämaa for technical assistance with cytometric bead array analysis.
Footnotes
-
Supported by the regional agreement on medical training and clinical research between Stockholm County Council and Karolinska Institutet, the Swedish Association Against Rheumatism, the Swedish Science Council, the Freemason Lodge Barnhuset in Stockholm, King Gustaf V’s Foundation, von Kantzow’s Foundation, Tore Nilsson’s Foundation, Swedish Society of Medicine, Jerring Foundation, the Gothenburg Medical Society, the Gothenburg Association against Rheumatism, Rune and Ulla Amlövs Foundation, St. Family Thölens and Kristlers Donations Foundation, Petter Silfverskiölds Foundation, the University of Gothenburg, Interdisciplinary Centre of Clinical Research at the University of Muenster (IZKF CRA04), and TARLA 0475 and DARLA 0500.
- Accepted for publication May 29, 2013.








{kind=link}
{kind=link}
{kind=link}
{kind=link}