
To the Editor:
A 57-year-old Iraqi woman presented with a 3-year history of right temporal headache, weight loss, and fatigue. She denied fevers, chills, night sweats, visual changes, jaw claudication, joint pain, or focal neurologic symptoms. Her medications included aspirin and captopril. She had received 1 month of oral prednisone (dose unclear) 4 months prior to presentation. Her vital signs were unremarkable, including symmetric blood pressures in all 4 limbs. She had scalp tenderness without temporal artery nodularity and had bruits of the right carotid, right brachial, and bilateral femoral arteries. The examination was otherwise unremarkable.
She was anemic (hematocrit 27.2%) with erythrocyte sedimentation rate (ESR) 115 mm/h and C-reactive protein (CRP) 3.3 mg/dl. Cerebrospinal fluid (CSF) was clear and colorless and showed a lymphocytic pleiocytosis (24 white blood cells with 93% lymphocytes) with mildly elevated protein (71 mg/dl) and normal glucose (51 mg/dl). Negative infectious studies of CSF included bacterial, fungal, and mycobacterial cultures, as well as venereal disease research laboratory test, PCR for herpes simplex virus, and cryptococcal antigen. Magnetic resonance imaging of the brain showed diffuse pachymeningeal thickening and enhancement (Figure 1A) without venous sinus thrombosis by computed tomography (CT) venogram. CT of the chest, abdomen, and pelvis revealed thoracic aortitis (Figure 1B).
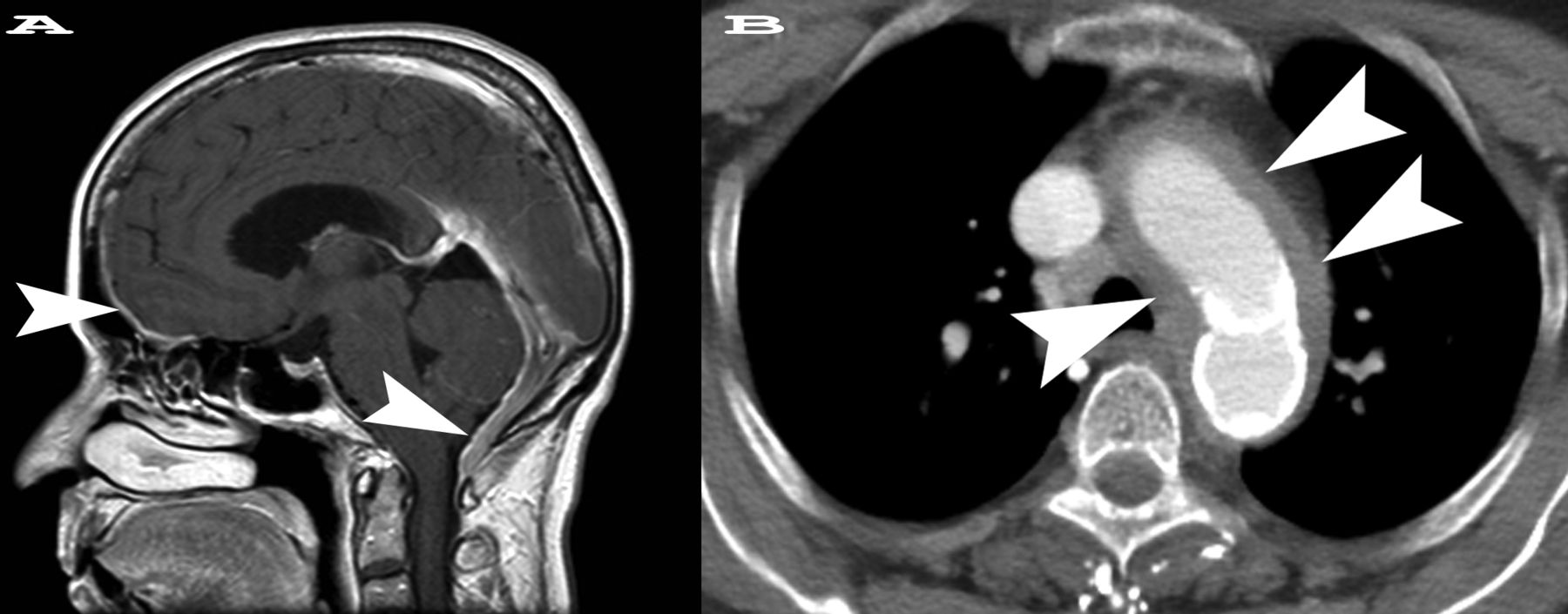
A. Sagittal postcontrast T1-weighted magnetic resonance scan shows diffuse pachymeningeal thickening and enhancement best seen in the anterior and posterior cranial fossa (arrowheads). The anterior cranial fossa arrowhead shows the future site of dural biopsy. The dural thickening had completely resolved on followup imaging 6 months after therapy was initiated. B. Axial postcontrast computed tomography image from the chest shows circumferential wall thickening surrounding the aortic arch and proximal great vessels (arrowheads) consistent with aortitis. There is a small amount of hyperattenuation in the aortic wall because of calcification. On followup imaging, the soft-tissue thickening was almost completely resolved.
Rheumatoid factor was 21 IU/ml. Serum antinuclear antibody, anti-neutrophil cytoplasmic antibody, interferon gamma release assay, rapid plasma reagent, and fluorescent treponemal antibody-absorbed tests were negative, and immunoglobulin G4 (IgG4) level was normal (54 mg/dl). The differential diagnosis included vasculitis (giant cell vs Takayasu arteritis) and IgG4-related disease (IgG4-RD), with sarcoidosis, malignancy, and infection considered less likely. A 20 × 2.5 mm biopsy of the right temporal artery showed no evidence of giant cell arteritis.
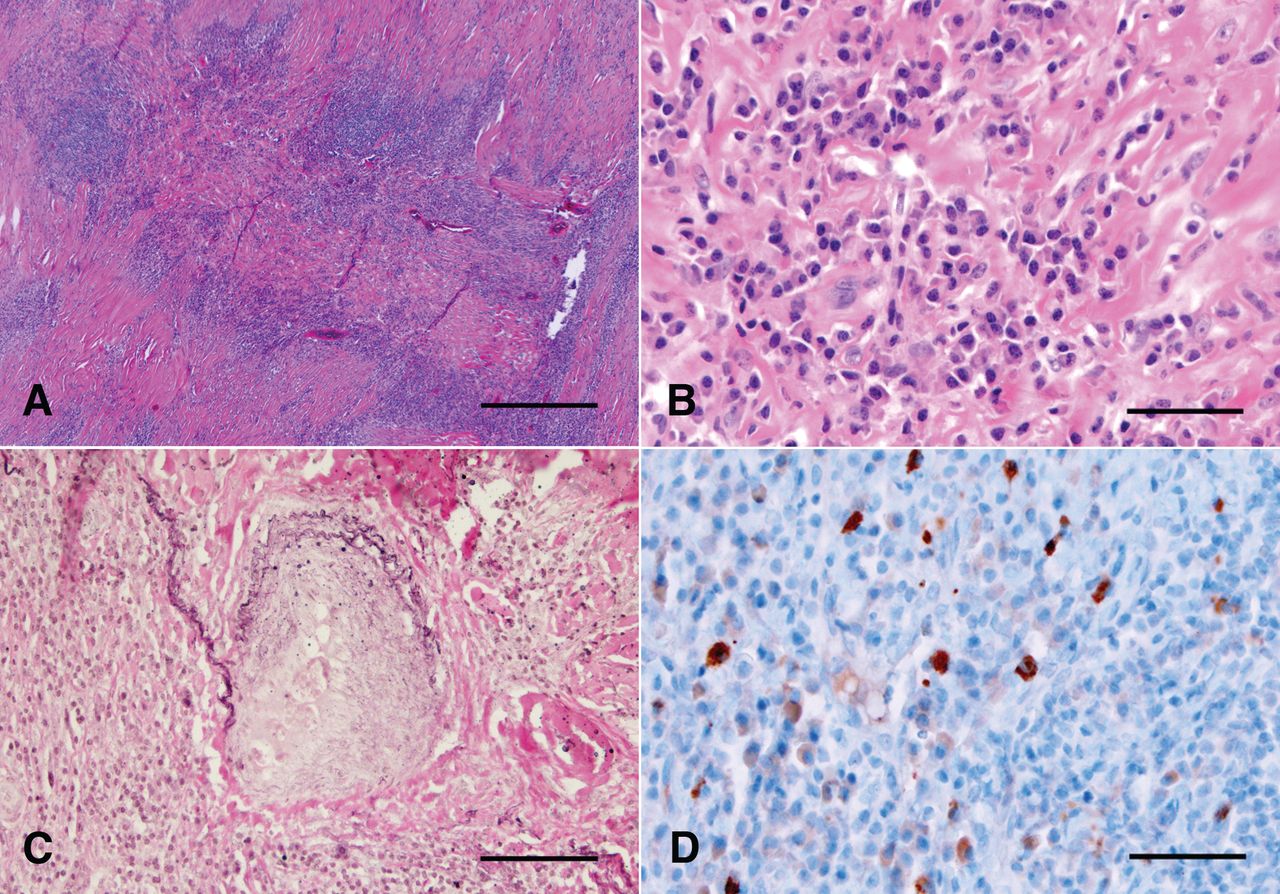
Dural biopsy demonstrated dural thickening with a dense lymphoplasmacytic infiltrate, lymphoid aggregates, focal storiform fibrosis, and focal phlebitis (Figure 2A–2C). There were rare eosinophils, abundant interspersed CD68-positive macrophages, and occasional giant cells, but no granulomas. Immunostaining for IgG4 and IgG revealed an elevated number of IgG4-positive plasma cells [average of 26 per high-power field (HPF); Figure 2D] and an IgG4/IgG ratio > 50%. Bacterial, fungal, and mycobacterial stains and cultures were negative and viral cytopathic changes absent. Immunohistochemistry supported an inflammatory process rather than lymphoma.

{kind=link}
{kind=link}
A. Low-power H&E stained section showing dense inflammatory infiltrate with lymphoid nodules. Bar = 500 μm. B. High-power H&E stained section showing plasma cell infiltrate and fibrosis. Bar = 50 μm. C. Elastin stain showing obliterative phlebitis. Bar = 100 μm. D. IgG4 immunostain highlights increased IgG4-positive plasma cells (in brown) within and adjacent to a lymphoid nodule. Bar = 50 μm.
IgG4-RD manifesting as pachymeningitis and aortitis was diagnosed. Prednisone was initiated at 0.6 mg/kg/day. Given concomitant severe osteoporosis, steroid-sparing agents in the form of weekly methotrexate and intravenous rituximab (1000 mg twice, 2 weeks apart) were administered 3 and 6 months from the time of diagnosis, respectively. Repeat CT imaging 6 months after initiation of prednisone showed complete resolution of the dural enhancement and marked improvement of the aortitis. Nine months from diagnosis, headaches had resolved and ESR (34 mm/h) and CRP (1.0 mg/dl) were markedly improved while she was taking prednisone 8 mg/day and weekly oral methotrexate. Repeat IgG4 level was 18 mg/dl.
IgG4-RD is a recently recognized fibroinflammatory entity characterized by tissue infiltration by IgG4-positive plasma cells1. It was first described in patients with autoimmune pancreatitis, in whom the presence of identical histological findings in other organs suggested a systemic process. It has since been described in myriad organs, with unifying histopathologic features2.
Pathogenesis of IgG4-RD remains unclear. Serum IgG4 level is elevated in 44%–100% of patients in published series1,3. A prozone effect caused by antigen excess may account for a subset of cases with reportedly normal IgG4 level, and may be overcome with serial sample dilutions3 (unfortunately not available in our patient). Histologically, a dense lymphoplasmacytic infiltrate in a storiform pattern is characteristic, yet not entirely specific. The abundant macrophages seen in this case are unusual for IgG4-RD in many tissues, but have been reported in meningeal disease4. For diagnosis, immunostaining must demonstrate an elevated absolute number of IgG4-positive plasma cells per HPF, and an elevated ratio of IgG4 to IgG-positive plasma cells may aid in confirmation. Mimics, particularly lymphomas, must be excluded1.
IgG4-RD has been found to cause a significant proportion of cases of noninfectious aortitis5. In contrast, IgG4-RD of the central nervous system is rare, with a few cases of pituitary and meningeal involvement described6. Recently, cases of both intracranial and spinal pachymeningitis due to IgG4-RD have been reported; these would previously have been classified as idiopathic hypertrophic pachymeningitis4,6,7. Consensus criteria for meningeal IgG4-RD would classify this case as “probable histological features of IgG4-RD” based on histological features and > 10 IgG4-positive cells per HPF8.
We are aware of 1 other case in the literature of IgG4-RD presenting with both aortitis and pachymeningitis9. In that case CSF IgG4 level was elevated and this may aid with diagnosis.
Challenges in the diagnosis of IgG4-RD include lack of widespread familiarity with the disease, absence of reliable diagnostic tests short of biopsy, protean manifestations, and many possible mimics. Glucocorticoids are the mainstay of therapy, although data increasingly support a role for rituximab for patients in whom steroids are contraindicated or ineffective10.
Acknowledgment
Special thanks to Dr. Marjorie Grafe, Division of Neuropathology, for her critical contributions.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.