

Abstract
Objective. Insights into the pathogenesis of inflammatory myopathies have led to new diagnostic methods. The aims of our study were (1) to evaluate the consequences of using the classification of Amato/European Neuromuscular Centre Workshop (ENMC) compared to that of Bohan and Peter; and (2) to evaluate any diagnostic benefit in using an extended pathological investigation.
Methods. From a consecutive retrospective database, we evaluated 99 patients for classification. Patients with inclusion body myositis (IBM) were classified according to Griggs, et al. In addition to routine stainings and immunohistochemistry, a multilevel serial sectioning procedure was performed on paraffin-embedded material, to identify scarce pathological findings.
Results. Classification according to Bohan and Peter could be performed for 83 of the 99 patients, whereas only 60 patients met the Amato/ENMC criteria, the latter resulting in the following diagnostic groups: IBM (n = 18), nonspecific myositis (n = 14), polymyositis (n = 12), dermatomyositis (n = 10), dermatomyositis sine dermatitis (n = 5), and immune-mediated necrotizing myopathy (n = 1). Most of the Amato/ENMC diagnostic groups harbored patients from several of the Bohan and Peter groups, which included a substantial group lacking proximal muscle weakness. The serial sectioning procedure was essential for classification of 9 patients (15%), and led to a more specific diagnosis for 13 patients (22%) according to Amato/ENMC.
Conclusion. The classification of Amato/ENMC was more restrictive, forming groups based on clinical criteria and specified myopathological findings, which clearly differed from the groups of the Bohan and Peter classification. An extended pathological investigation increased the diagnostic yield of a muscle biopsy and highlights the quantity and specificity of certain pathological findings.
- INFLAMMATORY MYOPATHIES
- IDIOPATHIC INFLAMMATORY MYOPATHIES
- POLYMYOSITIS
- DERMATOMYOSITIS
- INCLUSION BODY MYOSITIS
There have been new insights into the pathogenesis of the idiopathic inflammatory myopathies (IIM) since the classification of Bohan and Peter in 1975. New diagnostic entities, especially inclusion body myositis (IBM), have emerged1,2,3,4,5,6,7,8,9,10,11, and certain pathological findings have been closely tied to different IIM1,6,8,12. Many investigators have therefore advocated that this classification should be replaced13,14,15,16,17. A new classification developed by Amato for research purposes and clinical trials was modified and adopted at the European Neuromuscular Centre Workshop (ENMC) in 200314, referred to as the Amato/ENMC classification. Increased awareness of delayed diagnoses of IBM and overlap syndromes has also brought attention to the entity of “pure polymyositis”6,17,18, which was some years ago reported as uncommon19. The requirement for classification of specific pathological findings also focuses attention on the well known problem of uneven distribution of pathological processes in muscle, sometimes requiring repeat biopsies20. This issue has been addressed in studies concerning pathological criteria for IBM21, but not for partial invasion and perifascicular atrophy, as required for classification of polymyositis and dermatomyositis, respectively.
We evaluated the applicability and consequences of applying the Amato/ENMC classification to consecutive retrospective material of patients with a pathological diagnosis of inflammatory myopathy, and compared the outcome with use of the Bohan and Peter criteria. We also investigated the added value of a multilevel serial sectioning of biopsies for diagnosis of inflammatory myopathies.
MATERIALS AND METHODS
We searched the register of our muscle pathology laboratory from the years 1997 through 2002 for biopsies satisfying pathology criteria for inflammatory myopathy, i.e., showing inflammatory infiltrates and muscle fiber degeneration. In total, 164 patients were found; 120 of them could be identified with a current address in the national registry (see an overview of the inclusion process, Figure 1). Letters were sent to the 120 patients, and 99 (82.5%) responded and gave their consent. The medical records were scrutinized for relevant clinical, laboratory, and pathology data, using a standardized protocol. Information was gathered under specific headings: age, sex, date of biopsy, diagnostic code, heredity, social data, use of drugs, clinical examination, neuromuscular examination (especially strength testing), diagnosis of the treating physician, diagnoses of autoimmune disorders or other diseases, response to therapy, and further clinical course (including known or emerging malignancy, systemic inflammatory diseases, adverse drug effects, etc.). Because antibody testing was performed at different laboratories with different methods, only the antinuclear antibody (ANA) and extractable nuclear antigen (ENA) screen was documented. Immunofluorescence on HEp-2 cells was used in all cases for ANA screening, and ELISA was used in most cases for ENA screening. Muscle weakness as a criterion was understood to be a clearly detectable weakness, as judged by the examiner, also meeting the requirements as specified by the respective classification. As a response to treatment, only definite improvement in muscle strength was assessed, as either complete or near-complete improvement, obvious but not near-complete improvement, or less than that. In some patients no treatment was given. That the majority of patients with IBM were treated reflects a common delay of a definite diagnosis, and, at that time, a more optimistic attitude toward treatment of these patients than today. Muscle biopsy and clinical examination were done by ourselves in 44 of the 99 patients, but the medical records of all patients were thoroughly reviewed. In 44 of the patients the biopsy was taken from the anterior tibial muscle, in 21 from the lateral vastus, in 10 from the deltoid muscle, in 7 were from various muscles, and in 17 cases, the source of the muscle biopsy was not stated.

The process of selection of subjects. The laboratory analyzed 1807 biopsies during 1997–2002; 164 patients had biopsies showing inflammatory infiltrates and muscle fiber necrosis. Of 120 patients that were found in the national registry, 99 (82%) gave consent to have their data scrutinized. Sixteen patients did not have an inflammatory myopathy as defined by the Bohan and Peter, Amato/ENMC, or Griggs criteria. Eighteen patients had inclusion body myositis, 42 met the criteria of other inflammatory myopathies according to Amato/ENMC classification. The remaining 23 patients could be classified only according to Bohan and Peter. ENMC: European Neuromuscular Centre.
The study was approved by the regional ethical committee in Linköping.
The patients were classified according to Amato/ENMC and to Bohan and Peter criteria, and those with findings indicative of IBM were classified according to Griggs, et al10. In addition to systemic inflammatory diseases specifically required by Bohan and Peter, patients with mixed connective tissue disease and antisynthetase syndrome were diagnosed with an overlap syndrome, as judged by their treating rheumatologist. From each biopsy of all patients, there were, in addition to 24–30 sections of frozen tissue for histology, histochemistry, and immunohistochemistry (standard procedure), at least 2 pieces of muscle tissue collected, which were fixed in formalin and embedded in paraffin. These tissue pieces had been serially sectioned according to the following protocol (multiple-level, paraffin-embedded): 4 sections (5 μm thickness) were cut, 50 μm tissue was discarded, another 4 sections cut and another 50 μm tissue was discarded. This was repeated until at least 84 sections were available (extended procedure). The sections were stained with H&E (Ehrlich) alternated with Verhoeff-van Gieson (VG). In cases where MHC class I stainings were performed, the extent and pattern of expression was graded according to the previously described scale, where 1–5 signify increasing grades of expression22, and the number of cases was recorded in which the expression in the sarcolemma was widespread (thus supporting a diagnosis of IIM grade 3–5).
All biopsies were reevaluated by 1 investigator (OD) with regard to pathological findings relevant for classification. The reevaluation was blinded to the initial scrutiny, carried out several years ago, by another muscle pathologist (K. Henriksson, Neuromuscular Unit, Linköping University Hospital). The results were identical with regard to the presence of inflammatory infiltrates and muscle fiber degeneration, but in a few cases the presence of partial invasion was not specifically stated in the original investigation. All biopsies in which at least 1 partial invasion had been detected in the extended screening (multilevel serial sectioning) now underwent a second serial sectioning procedure of frozen tissue (single-level sequential). This procedure was performed according to the following description: 35 subsequent sections of each biopsy were collected and indexed in the following manner: 1a1, 1a2, 1a3, 1b, 1c1, 1c2, 1c3, 2a1,..., 5c3. The b-sections were 8 μm thick and the a- and c-sections were 6 μm. The b-sections were stained with H&E. Where the morphology of H&E-stained sections showed a partial invasion, the preceding or subsequent 3 sections were stained in the following manner: the 1-sections with antibodies against MHC I [HLA-ABC antigen, clone W6/32(1)], the 2-sections with antibodies against CD8 (CD8, clone DK25), and the 3-sections with antibodies against MAC (C5b-9, clone aE11), all from Dako Denmark A/S. The primary antibodies were visualized with a detection kit including enzyme-conjugated secondary antibodies supplied by the manufacturer (Starr Trek Universal HRP, Biocare Medical). An additional similar procedure was undertaken with 17 biopsies suggestive of IBM, where the sections were all 10 μm thick and the b-sections were stained with modified Gomori trichrome23 and the a- and c-sections were stained with alkaline Congo red according to Puchtler, et al24. Earlier sections of these biopsies had shown changes suggestive of, but not diagnostic of IBM (no amyloid seen in 3 random sections stained with Congo red). The added value of the multilevel serial sectioning to detect and classify an inflammatory myopathy and the single-level sectioning to diagnose IBM, respectively, was calculated.
RESULTS
Population characteristics, laboratory findings, association with other systemic inflammatory disorders or neoplasia and treatment response are shown in Table 1.
Characteristics of the study population including demographic, laboratory, and clinical data.
Of the 99 patients included in the study, 60 met the criteria of Amato/ENMC or Griggs, while an additional 23 patients could be classified according to Bohan and Peter or Griggs. Thus, in total 83 patients were classified. Of the remaining 16 patients, those excluded from both classifications, there were 6 patients who just met the pathology criteria for inflammatory myopathy, 3 who were found to have a muscular dystrophy, 2 who had taken myotoxic drugs (statins), 2 in whom the weakness could be explained by other diseases (motor neuron disease and neuroborreliosis, respectively), 1 considered to have rhabdomyolysis, 1 with granulomatous disease without weakness, and 1 patient where data needed for classification were not considered sufficient.
The largest group classified according to Amato/ENMC/Griggs was that of patients satisfying Griggs criteria for IBM (n = 18; 30%; definite/possible 14/4). The second largest group had nonspecific myositis (n = 14; 23%), followed by the group with polymyositis (n = 12; 20%; definite/probable 11/1) and those with dermatomyositis (n = 10; 17%; definite/probable 6/4). The groups with possible dermatomyositis sine dermatitis (n = 5) and immune-mediated necrotizing myopathy (n = 1) were smaller (Figure 2). Of the additional 23 patients meeting the criteria of Bohan and Peter, 21 lacked a detectable proximal weakness, which is a mandatory criterion according to Amato/ENMC, and for 2 patients there were no other specified laboratory criteria, which had been required for inclusion in these 2 cases. Electromyography (EMG) and magnetic resonance imaging (MRI) were not performed and only a few myositis-specific antibodies (MSA) were investigated in these 2 particular cases. Classification according to Bohan and Peter resulted in the following groups: polymyositis (definite, probable, possible, 14/5/7, respectively per group), dermatomyositis (2/2/1), IIM associated with cancer (4/1/0), childhood IIM (2/0/0), and overlap syndrome (10/13/4). In 74 of the 83 classified cases, MHC class I staining was performed, and showed in 72 cases widespread expression in the sarcolemma (grade 3–5), clearly supporting the diagnosis. One of the 2 cases with weaker MHC class I expression had been exposed to steroids prior to biopsy and in the other case the diagnosis had otherwise strong support.
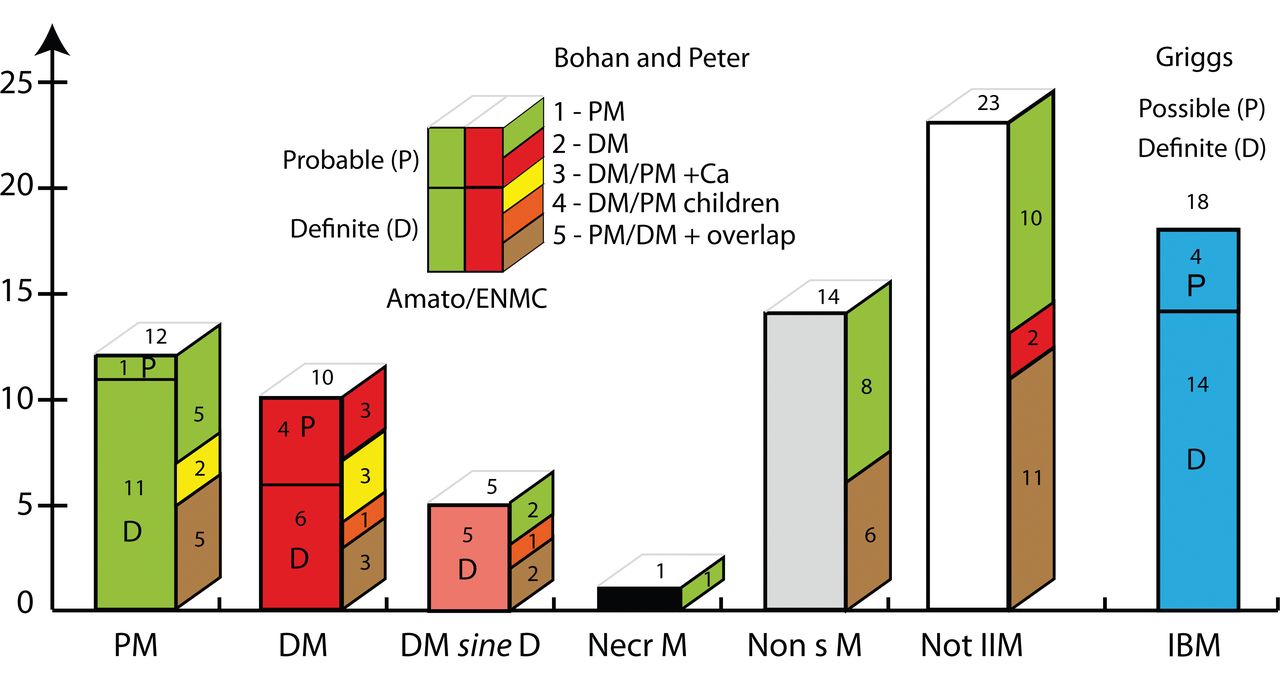
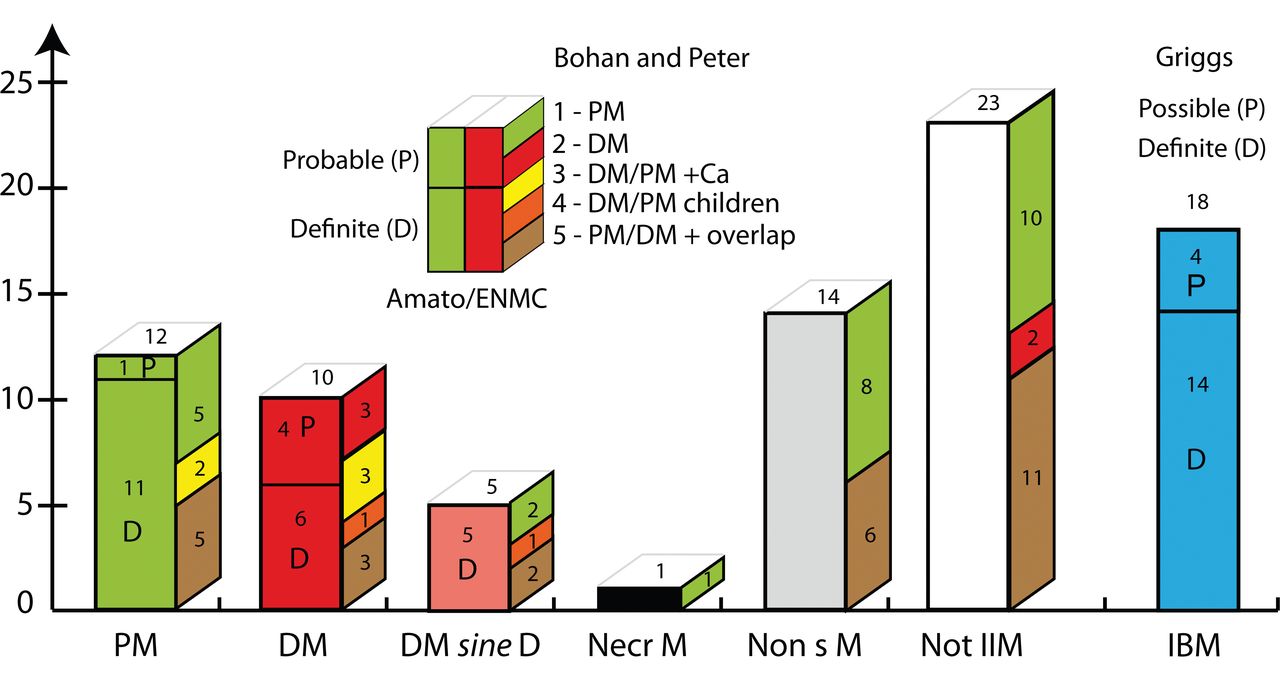
The diagnostic groups. Front side of bars shows numbers of patients diagnosed according to the classification of Amato/ENMC, and the lateral side shows the corresponding numbers of patients in these groups classified according to Bohan and Peter. Patients with definite or probable inclusion body myositis (IBM) are classified according to Griggs and are displayed in a separate bar. The insert explains the principle that cases classified as polymyositis (PM) and dermatomyositis (DM) according to Amato/ENMC (front side) are divided into definite (D) and probable (P) and that the color coding on the lateral side shows the classification groups (given by numbers) according to Bohan and Peter. The Bohan and Peter groups 3, 4, and 5 [patients with associated cancer (Ca), children with IIM, or overlap syndromes] could be viewed as a subgroup within the Amato/ENMC classification by adding these designations. Details of classifications are given in the Appendix. DM sine D: possible dermatomyositis sine dermatitis; Necr M: immune-mediated necrotizing myopathy; Non sM: nonspecific myositis; Not IIM: not classified as an idiopathic inflammatory myopathy according to Amato/ENMC (but according to Bohan and Peter); IBM: inclusion body myositis according to Griggs. Insert: PM: polymyositis; DM: dermatomyositis (adults); DM/PM+Ca: DM or PM with neoplasia (cancer; within 4 years from biopsy); DM/PM children: childhood DM or PM associated with vasculitis; PM/DM + overlap: PM or DM associated with collagen vascular disease. ENMC: European Neuromuscular Centre.
Looking into details of differences and similarities in the classified patients (Figure 2), we found that of the 12 patients classified as polymyositis according to Amato/ENMC, 5 were also polymyositis according to Bohan and Peter, while with the latter classification another 2 were designated with an association with cancer (within 4 years from biopsy) and 5 were diagnosed with an overlap syndrome. Of the 10 patients with dermatomyositis according to Amato/ENMC, 3 were also diagnosed as dermatomyositis according to Bohan and Peter, 3 as IIM associated with cancer, 3 as an overlap syndrome, and 1 with childhood IIM. In the group of 5 patients classified by Amato/ENMC as possible dermatomyositis sine dermatitis, 2 were classified as polymyositis, 2 as overlap syndrome, and 1 with childhood IIM, according to Bohan and Peter.
The group of nonspecific myositis in the Amato/ENMC classification, i.e., a group of IIM lacking the specific criteria needed for classification into other specified groups, comprised 14 patients. Of these, 8 were designated polymyositis and 6 overlap syndrome according to Bohan and Peter. The 23 patients that were not classified as IIM according to Amato/ENMC were diagnosed as polymyositis (n = 10), overlap syndrome (n = 11), or dermatomyositis (n = 2) according to Bohan and Peter. We chose to present patients meeting the criteria for IBM as a separate entity, next to the other diagnostic groups, in both classifications (Figure 2).
When the results with and without the use of multiple-level serial sectioning were compared, using the Amato/ENMC classification this procedure allowed the inclusion of a further 9 patients (15%) where the diagnosis of inflammatory myopathy had otherwise been overlooked, and led to a more specific diagnosis (e.g., definite instead of probable) in 13 patients (22%). Using the Bohan and Peter classification (now without the IBM patients), the multilevel serial sectioning detected findings essential for inclusion in 7 additional patients (11%), and allowed a more specific diagnosis in 15 (23%). When single-level serial sectioning using the protocol above with different immunohistological stainings was undertaken, at least 1 partial invasion meeting the Amato/ENMC criteria could be confirmed in biopsies from 36 of the 83 classified patients. The described single-level (sequential) serial sectioning also turned out to be a valuable tool to detect amyloid material, allowing the diagnosis of definite IBM in another 9 IBM patients. An example of partial invasion of CD8-positive cytotoxic T cells, in a non-necrotic muscle fiber upregulating MHC I, and a muscle fiber with rimmed vacuoles and amyloid substance are shown in Figure 3.
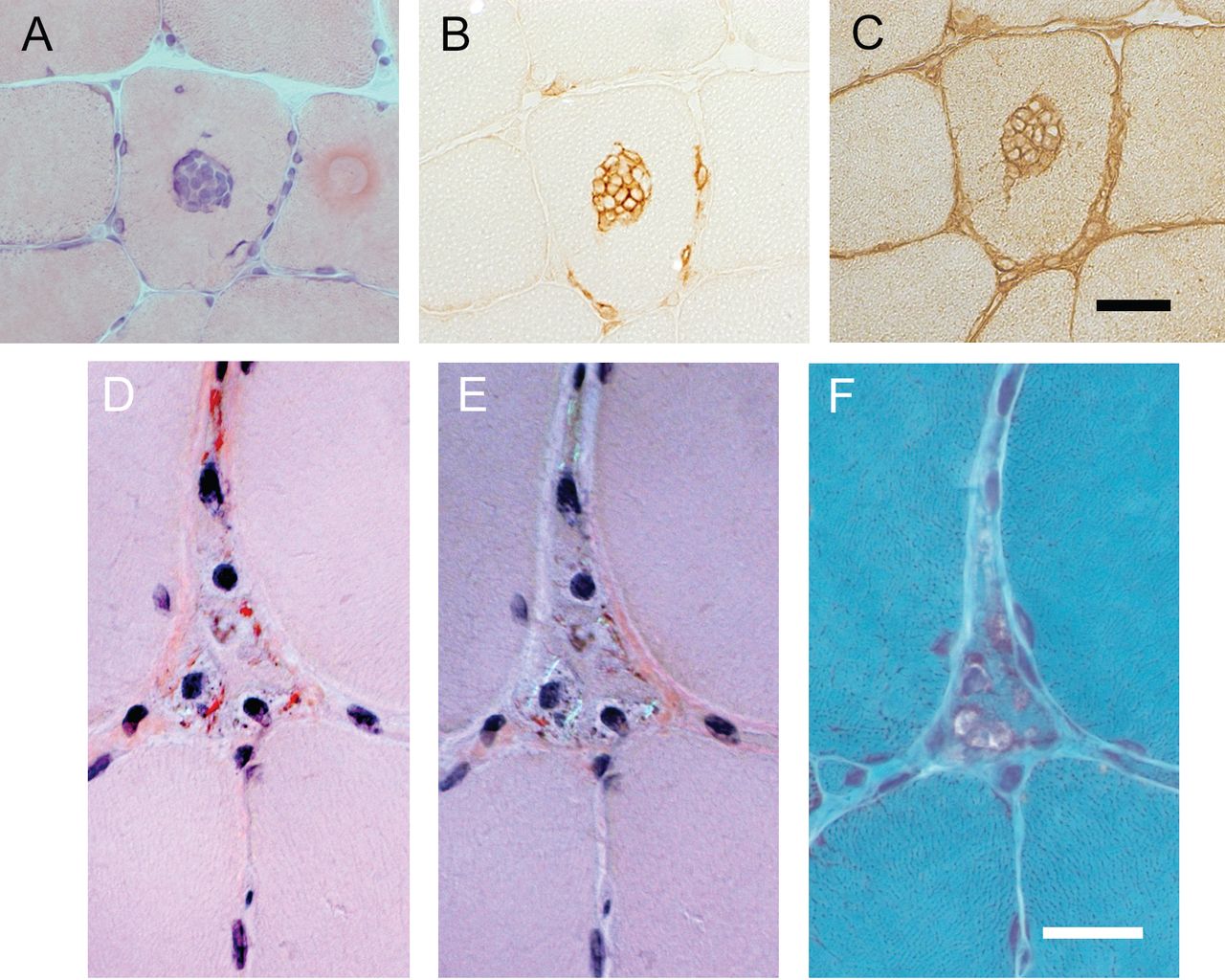
Partial invasion in 1 fiber and amyloid in a fiber with vacuoles. A. Typical morphological finding of partial invasion (H&E stain). B and C, respectively: Immunohistological staining for CD8 and MHC I (C) shows partial invasion of CD8-positive cytotoxic T cells in a non-necrotic muscle fiber, upregulating MHC I. The sections were adjacent on serial sectioning. D. A fiber with amyloid deposits (Congo red stain), also visualized with a turned polarized lens (E), contains rimmed vacuoles (Gomori trichrome stain; (F) on serial sequential sectioning. Bars are all 20 μm.
Concerning specific pathological findings highlighted in the Amato/ENMC criteria, the 36 patients showing partial invasion in their biopsy results comprised not just patients with IBM or polymyositis, but also 2 cases with a possible dermatomyositis sine dermatitis, 1 with definite dermatomyositis, and 6 patients who had no detectable weakness, the latter group not allowing classification according to Amato/ENMC. Four of these 6 patients without weakness had polymyositis according to Bohan and Peter and 2 had an overlap syndrome, adding up to 8 patients with partial invasions diagnosed with an overlap syndrome. In all the biopsies showing a partial invasion in histological staining, this could also be confirmed with sequential immunohistological sections, except in 1 case (probably due to technical problems as the tissue was damaged), indicating that immunohistological confirmation may not be necessary, if the morphology is straightforward. Biopsies from 15 of the classified patients (n = 83) showed perifascicular atrophy; of these, 6 patients had dermatomyositis, 5 had possible dermatomyositis sine dermatitis, and 4 were not eligible for inclusion according to the Amato/ENMC criteria. Biopsies from 4 patients with perifascicular atrophy also showed partial invasion. One of these patients had an otherwise classical dermatomyositis associated with breast cancer (biopsy section shown in Figure 4), 2 had possible dermatomyositis sine dermatitis, and 1 patient did not meet the Amato/ENMC criteria.
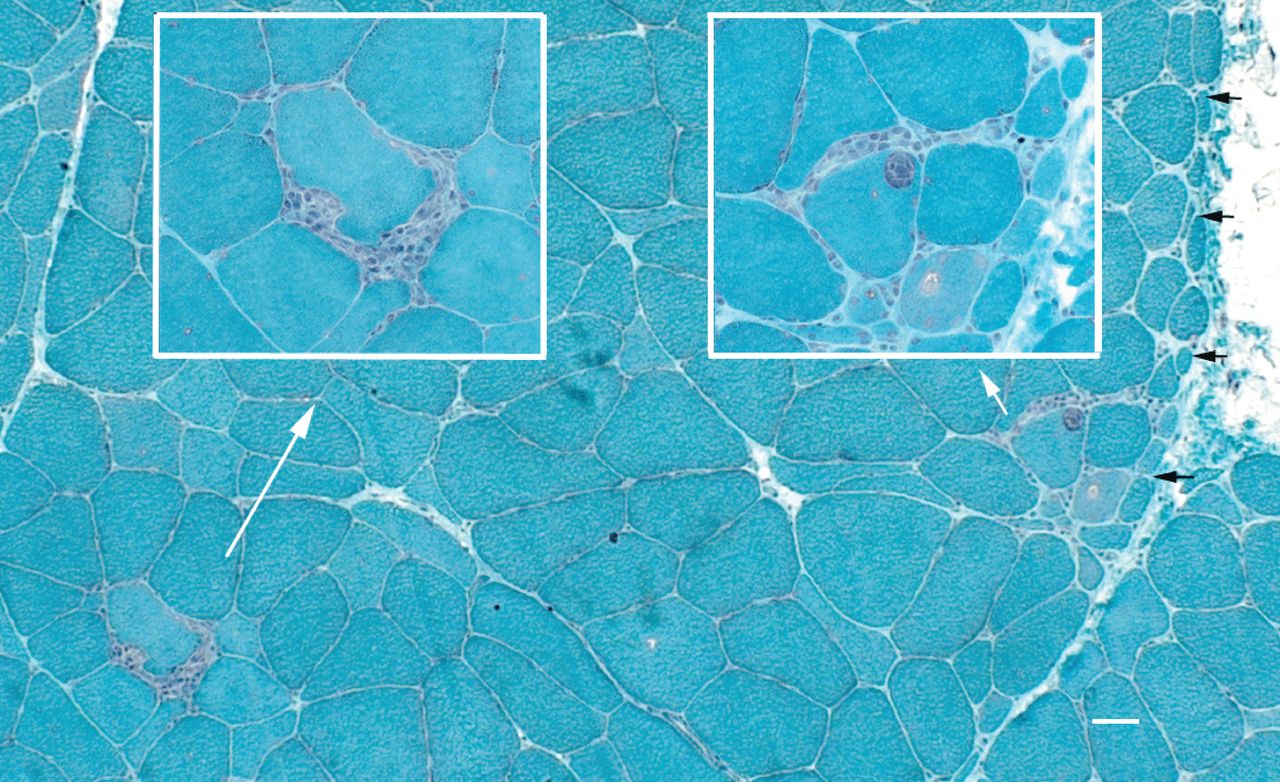
Concomitant findings of perifascicular atrophy and partial invasions in 1 biopsy section, from a patient with classical dermatomyositis with an associated neoplasia. White arrows show the origin of the insert panels with higher magnifications. Black arrows mark the area of perifascicular atrophy. Bar = 20 μm.
DISCUSSION
The Amato/ENMC classification has taken much of the recent concepts of pathogenesis and pathology into account, and was recommended by the 119th European Neuromuscular Centre (ENMC) international workshop for future randomized therapeutic studies14. We have used the Amato/ENMC classification in consecutive material of muscle biopsies and compared it to the classification of Bohan and Peter. The Griggs classification of IBM that emerged after the Bohan and Peter criteria, also referred to in the Amato/ENMC classification, is now widely accepted, and consequently was used here14,25.
IBM was the most common and polymyositis the next most common diagnosis of the defined IIM when applying the Amato/ENMC criteria in this population. Thus, IIM with typical pathological hallmarks of polymyositis was not a particularly rare entity in relation to other IIM, in accord with results from Chahin and Engel18. However, our data show that partial invasion as defined by Amato/ENMC was also present in muscle from several patients with associated systemic inflammatory disorders or patients without weakness, who had pathological findings consistent with an inflammatory myopathy, indicating that this pathoimmunological process may not be so disease-specific as previously thought.
One major difference between the classifications is that the Bohan and Peter criteria specifically create groups for patients having another systemic inflammatory disease (overlap syndromes), or a cancer, disorders that may be pathogenetically linked to an inflammatory myopathy, whereas the Amato/ENMC acknowledges designation of these associated disorders within the defined groups. An overlap syndrome was present in 42% and 30%, and an associated cancer was present in 17% and 30%, respectively, of the Amato/ENMC classified polymyositis and dermatomyositis groups.
Another major difference between the classifications is that the Amato/ENMC classification creates groups that are not specifically classified. The first group, nonspecific IIM, i.e., myositis, lacking specific pathologic criteria, comprised patients classified as polymyositis or overlap syndrome according to Bohan and Peter. The second group that was not specifically classified according to Amato/ENMC designated patients as not having IIM, the main reason (applicable in 21 of 23 patients) being the lack of proximal muscle weakness, which is required for classification according to Amato/ENMC. Designed specifically for research purposes, including therapeutic trials, the Amato/ENMC classification would make sense in this respect by avoiding patients lacking muscle weakness. However, many patients without a detectable weakness who exhibit myopathological findings and inflammatory infiltrates also show laboratory signs indicating an ongoing destructive muscle disease. Several of these patients also present with a systemic inflammatory disease needing treatment, while others have a generalized myalgia, in accord with considerations from Bohan and Peter, motivating their fifth diagnostic group for overlap syndromes, and inclusion of patients with lower degrees of diagnostic certainty4.
There is often a sampling problem in muscle biopsy diagnosis, because many pathological processes are focally distributed, and 1 isolated finding is not often proof of a certain disease20. This aspect may be especially important for classifications emphasizing specific pathological findings. The method of multilevel serial sectioning described in this report increased the benefit of each biopsy to detect and classify IIM according to the criteria established by Amato/ENMC and Griggs, as well as by Bohan and Peter. The introduction of immunohistochemistry, including MHC class I expression, has added another useful tool for diagnosing IIM26,27, although an upregulation of MHC class I is also seen in some dystrophies, and the extent of expression is variable and is sometimes also moderately upregulated in healthy controls, making standardizations between laboratories and cutoff values necessary6,26,27,28.
Notably, our results indicate that pathologic findings considered more or less specific for certain diseases are also found in muscle biopsies from patients with other disease processes, which also was evident in the comprehensive quantitative descriptions given by Arahata and Engel1. Therefore, the frequency of different findings should probably be considered, and consequently the question is raised of how extensive the pathological investigation should be when classifying patients, not only in a clinical situation but also in the context of therapeutic trials. Our findings also signal some caution not to rely too much on any specific criterion, that being partial invasion, perifascicular atrophy, or the localization of an inflammatory infiltrate. This is also in accord with previous investigations of patient cohorts in our unit29, and indicates that, until more quantitative data are available, a classification that separates the pathological findings from the clinical syndromes may be an attractive alternative, as recently suggested by Pestronk30.
Limitations of our study are those inherent with retrospective data, and the data are not valid as conclusive epidemiological results other than that the number of patients in each group as stated represents a minimum in our referral area. The use of strict pathological findings as screening criteria as well as using semiquantitative evaluation of MHC class I expression would be expected to reduce misdiagnoses, such as with false positives. However, some patients with IIM could have been disregarded if a biopsy was not carried out (considered rare in our referral area). Further, extensive immunohistochemical staining, Western blot, or genetic testing was performed in only a few cases when suspicion of muscular dystrophy arose. The amyopathic dermatomyositis diagnostic group did not meet our inclusion criteria, and it is not considered in our study. Similarly, patients with immune-mediated necrotizing myopathy may have been excluded (if no inflammation at all was present), thereby underestimating the frequency of this entity, which has drawn much attention recently31,32. Further, we investigated and examined fewer than half of the patients ourselves, which makes the study liable to bias for using secondary data, and the presence or absence of scapular winging, muscle hypertrophy, or atrophy was sometimes not explicitly commented on in the notes that were retrospectively reviewed. However, detailed neuromuscular examinations were recorded in the majority of cases, and to compensate for some of the uncertainties, medical records were repeatedly reviewed during a long followup time (median 7 years) and missing data were vigorously pursued, making misclassifications less likely. We did not investigate the shortcomings or merits of the previously widely used Bohan and Peter classification set. We took into account 2 of its most obvious problems for use in research studies, i.e., not requiring a muscle biopsy for probable or possible diagnoses and the latter emergence of IBM. Another important advance in the understanding of IIM that we did not address is the emergence of antibodies with a high specificity for subgroups of IIM.
We conclude that most patients screened by pathological findings (inflammation and fiber degeneration) who met the Bohan and Peter criteria could also be classified according to the Amato/ENMC classification, except a rather large group of patients lacking clearly detectable muscle weakness. Despite the strict definition according to the Amato/ENMC, polymyositis was not a particularly uncommon disease, constituting 20% of the classified patients, and it was surpassed in frequency only by the IBM (30%) and nonspecific myositis (23%) groups in our population. The main differences between the classifications are that (1) the Amato/ENMC classification creates smaller and more homogeneous groups of specific pathological findings, although some of these findings could also be found in other patients; (2) the Amato/ENMC classification creates groups with nonspecific IIM and non-IIM, groups that may need particular considerations in therapeutic trials; and (3) the Bohan and Peter classification denotes a substantial number of patients with overlap syndrome or associated cancer, as well as a childhood form of IIM, as separate groups, whereas the Amato/ENMC classification recognizes these patients within otherwise defined groups. Further, our data give definite support for the added value of a multilevel serial sectioning procedure, as an extended pathological method to diagnose and classify inflammatory myopathies.
Acknowledgment
Special thanks to Gunnvor Sjöö, Liv Gröntoft, and Bo Häggqvist for skillful technical assistance, and Karl Henriksson for introducing the method of multilevel serial sectioning and setting high standards.
Appendix
The classifications of Bohan and Peter, Amato/ENMC, and Griggs
Classification of idiopathic inflammatory myopathies according to Bohan and Peter1,2
Diagnostic groups-
Group I. Primary idiopathic polymyositis
-
Group II. Primary idiopathic dermatomyositis
-
Group III. Dermatomyositis (or polymyositis) associated with neoplasia
-
Group IV. Childhood dermatomyositis associated with vasculitis
-
Group V. Polymyositis and dermatomyositis with associated systemic inflammatory disease
-
Symmetrical weakness of the limb girdle muscles and neck flexor muscles progressing over weeks to months
-
Muscle biopsy evidence of necrosis of Type I and II fibers, phagocytosis, regeneration with basophilia, large vesicular sarcolemmal nuclei and prominent nucleoli, atrophy in a perifascicular distribution, variation in fiber size, and an inflammatory exsudate, often perivascular
-
Elevation in serum of skeletal muscle enzymes, particularly creatine phosphokinase and often aldolase, serum glutamate oxaloacetate, pyruvate transaminases, and lactate dehydrogenase
-
Electromyographic triad of short, small polyphasic motor units, fibrillations, positive sharp waves and insertional irritability, and bizarre, high frequency repetitive discharges
-
Dermatologic features including a lilac discoloration of the eyelids (heliotrope) with periorbital edema, a scaly, erythematous dermatitis over the dorsum of the hands (especially the metacarpophalangeal and proximal interphalangeal joints, Gottron’s sign), and involvement of the knees, elbows and medial malleoli, as well as face neck and upper torso
Confidence limits: Dermatomyositis: 3 or 4 of 1–4, plus 5 for definite, subtracting 1 or 2 of 1–4, for probable and possible respectively; Polymyositis: 4 of 1–4 for definite, subtracting 1 or 2 of 1–4, for probable and possible, respectively.
Exclusion criteria:-
Evidence of central or peripheral neurologic disease
-
Muscle weakness with a slowly progressive, unremitting course and a positive family history of calf enlargement to suggest a muscular dystrophy
-
Biopsy evidence of granulomatous myositis such as with sarcoidosis
-
Infections, including trichinosis, schistosomiasis, trypanosomiasis, staphylococcosis and toxoplasmosis
-
Recent use of various drugs and toxins, such as clofibrate and alcohol
-
Rhabdomyolysis as related to gross myoglobolinemia (several non-inflammatory causes are listed)
-
Metabolic disorders such as McArdle’s syndrome
-
Endocrinopathies (several mentioned)
-
Myasthenia gravis
-
Miscellaneous (several uncommon differential diagnoses are mentioned, virus-like particles observed in electron microscopy by Chou and Sato, later to be identified as characteristic tubulofilaments of IBM, are mentioned as a not yet settled issue, in another part of the article3,4,5,6
Classification for inflammatory myopathies according to Amato/ENMC7
-
Diagnostic groups
-
Polymyositis
-
Definite polymyositis
-
All clinical criteria with the exception of rash
-
Elevated CK
-
Muscle biopsy criteria include a, and exclude c, d, h, i
-
-
Probable polymyositis
-
All clinical criteria with the exception of rash
-
Elevated CK
-
Other laboratory criteria (1 of 3)
-
Muscle biopsy criteria include b, and exclude c, d, g, h, i
-
-
-
Dermatomyositis
-
Definite dermatomyositis
-
All clinical criteria
-
Muscle biopsy criteria include
-
-
Probable dermatomyositis
-
All clinical criteria
-
Muscle biopsy criteria include d or e, or elevated serum CK, or other laboratory criteria (1 of 3)
-
-
-
Amyopathic dermatomyositis
-
Rash typical of DM: heliotrope, periorbital edema, Gottron’s papules/sign, V-sign, shawl sign, holster sign
-
Skin biopsy demonstrates a reduced capillary density, deposition of MAC on small blood vessels along the dermal-epidermal junction, and variable keratinocyte decoration for MAC
-
No objective weakness
-
Normal serum CK
-
Normal EMG
-
Muscle biopsy, if done, does not reveal features compatible with definite or probable DM
-
-
Possible dermatomyositis sine dermatitis
-
All clinical criteria with the exception of rash
-
Elevated serum CK
-
Other laboratory criteria (1 of 3)
-
Muscle biopsy criteria include c or d
-
-
Nonspecific myositis
-
All clinical criteria with the exception of rash
-
Elevated serum CK
-
Other laboratory criteria (1 of 3)
-
Muscle biopsy criteria include e or f, and exclude all others
-
-
-
Immune-mediated necrotizing myopathy
-
All clinical criteria with the exception of rash
-
Elevated serum CK
-
Other laboratory criteria (1 of 3)
-
Muscle biopsy criteria include g, and exclude all others
-
-
Clinical criteria
-
Inclusion criteria
-
Onset usually over 18 years (post puberty), onset may be in childhood in DM and nonspecific myositis
-
Subacute or insidious onset
-
Pattern of weakness: symmetric, proximal > distal, neck flexor > neck extensor
-
Rash typical of DM: heliotrope (purple) periorbital edema; violacious papules (Gottron’s papules) or macules (Gottron’s sign), scaly if chronic, at metacarpophalangeal and interphalangeal joints and other bony prominences; erythema of chest and neck (V-sign) and upper back (shawl sign)
-
-
Exclusion criteria
-
Clinical features of IBM (see Griggs et al. (Ann Neurol 1995;38:705–13): asymmetric weakness, wrist/finger flexors same or worse than deltoids; knee extensors and/or ankle dorsiflexors same or worse than hip flexors)
-
Ocular weakness, isolated dysarthria, neck extensor > neck flexor weakness
-
Toxic myopathy (e.g., recent exposure to myotoxic drugs), active endocrinopathy (hyper- or hypothyroid, hyperparathyroid), amyloidosis, family history of muscular dystrophy or proximal motor neuropathies (e.g., SMA)
-
-
-
Elevated serum creatine kinase level
-
Other laboratory criteria.
-
Electromyography
Inclusion criteria: (I) Increased insertional and spontaneous activity in the form of fibrillation potentials, positive sharp waves, or complex repetitive discharges; (II) Morphometric analysis reveals the presence of short duration, small amplitude, polyphasic MUAPs
Exclusion criteria: (I) Myotonic discharges that would suggest proximal myotonic dystrophy or other channelopathy; (II) Morphometric analysis reveals predominantly long duration, large amplitude MUAPs; (III) Decreased recruitment pattern of MUAPs
-
MRI: diffuse or patchy increased signal (edema) within muscle tissue on STIR images
-
Myositis-specific antibodies detected in serum
-
-
Muscle biopsy inclusion and exclusion criteria
-
Endomysial inflammatory cell infiltrate (T cells) surrounding and invading non-necrotic muscle fibers
-
Endomysial CD8+ T cells surrounding, but not definitely invading non-necrotic muscle fibers, or ubiquitous MHC-I expression
-
Perifascicular atrophy
-
MAC deposition on small blood vessels, or reduced capillary density, or tubuloreticular inclusions in endothelial cells on EM, or MHC-I expression on perifascicular fibers
-
Perivascular, perimysial inflammatory cell infiltrate
-
Scattered endomysial CD8+ T-cells infiltrate that does not clearly surround or invade muscle fibers
-
Many necrotic muscle fibers as the predominant abnormal histological feature. Inflammatory cells are sparse or only slight perivascular; perimysial infiltrate is not evident. MAC deposition on small blood vessels or pipestem capillaries on EM may be seen, but tubuloreticular inclusions in endothelial cells are uncommon or not evident
-
Rimmed vacuoles, ragged red fibers, cytochrome oxidase-negative fibers that would suggest IBM
-
MAC deposition on the sarcolemma of non-necrotic fibers and other indications of muscular dystrophies with immunopathology
-
Classification of inclusion body myositis according to Griggs, et al8 [Developed by J. Mendell, R. Barohn, V. Askanas, M. Dalakas, S. DiMauro, A. Engel, G. Karpati, L.P. Rowland]
-
Characteristic features — inclusion criteria
-
Clinical features
-
Duration of illness > 6 months
-
Age of onset > 30 years old
-
Muscle weakness
Must affect proximal and distal muscles of arms and legs and patient must exhibit at least one of the following features: (a) Finger flexor weakness; (b) Wrist flexor > wrist extensor weakness; (c) Quadriceps muscle weakness (≤ grade 4 MRC)
-
-
Laboratory features
-
Serum creatine kinase < 12 times normal
-
Muscle biopsy
-
Inflammatory myopathy characterized by mononuclear cell invasion on non-necrotic fibers
-
Vacuolated muscle fibers
-
Either (i) Intracellular amyloid deposits (must use fluorescent method of identification before excluding the presence of amyloid; or (ii) 15–18 nm tubulofilaments by electron microscopy
-
-
Electromyography must be consistent with features of an inflammatory myopathy (however, long-duration potentials are commonly observed and do not exclude diagnosis of sporadic inclusion body myositis).
2. Family history: Rarely, inclusion body myositis may be observed in families. This condition is different from hereditary inclusion body myopathy without inflammation. The diagnosis of familial inclusion body myositis requires specific documentation of the inflammatory component by muscle biopsy in addition to vacuolated muscle fibers, intracellular (within muscle fibers) amyloid, and 15–18 nm tubulofilaments.
-
II. Associated disorders: Inclusion body myositis occurs with a variety of other, especially immune-mediated conditions. An associated condition does not preclude a diagnosis of inclusion body myositis if diagnostic criteria (below) are fulfilled.
III. Diagnostic criteria for inclusion body myositis
-
Definite inclusion body myositis: Patients must exhibit all muscle biopsy features including invasion of non-necrotic fibers by mononuclear cells, vacuolated muscle fibers, and intracellular (within muscle fibers) amyloid deposits or 15–18 nm tubulofilaments.
None of the other clinical or laboratory features are mandatory if muscle biopsy features are diagnostic.
-
Possible inclusion body myositis: If the muscle shows only inflammation (invasion of non-necrotic muscle fibers by mononuclear cells) without other pathological features of inclusion body myositis, then a diagnosis of possible inclusion body myositis can be given if the patient exhibits the characteristic clinical (Al, 2, 3) and laboratory (Bl, 3) features.
-
Footnotes
-
Supported by the University Hospital Linköping and the County Council of Östergötland.
- Accepted for publication February 14, 2013.








{kind=link}
{kind=link}
{kind=link}
{kind=link}