

To the Editor:
An 11-year-old girl was diagnosed with systemic juvenile idiopathic arthritis (sJIA) after presenting with prolonged fever, lymphadenopathy, arthritis, classic sJIA rash, and hyperferritinemia. Complicating macrophage activation syndrome (MAS) was diagnosed, noting hemophagocytosis on bone marrow biopsy for further evaluation of mental status changes. Despite high-dose glucocorticoids and cyclosporine, she remained febrile until the addition of daily subcutaneous interleukin 1 (IL-1) receptor antagonist anakinra (100 mg).
She later developed a methicillin-resistant Staphylococcus aureus abscess, and anakinra was withheld. After several days of intravenous antibiotics, she was discharged while taking oral antibiotics, prednisone, and cyclosporine. After anakinra had been withheld for 14 days, she was rehospitalized, this time for a flare of her sJIA, with fever, hypotension, and doubling of her ferritin level (9000 ng/ml). Daily anakinra was resumed, and she received solumedrol pulse therapy and intravenous cyclosporine for persistent fever and hyperferritinemia (peaking at 100,000 ng/ml). She was eventually discharged while taking cyclosporine, prednisone, and anakinra. She remained clinically quiescent for several weeks without fever and her serum ferritin decreased (300 ng/ml).
However, because she developed significant medication toxicity (weight gain, hypertension, hirsuitism, and paresthesias), her corticosteroid dose was tapered. She was soon readmitted with anorexia, malaise, and fever. Her medications on admission included prednisone 40 mg daily (0.7 mg/kg), cyclosporine 100 mg twice daily (3.2 mg/kg/day), and anakinra 100 mg daily (1.75 mg/kg). Her laboratory evaluation was significant for the following: ferritin 2456 ng/ml; C-reactive protein (CRP) 78.16 mg/dl; thrombocytopenia 120,000 platelets/mm3; hypertriglyceridemia 338 mg/dl; and erythrocyte sedimentation rate 3 mm/h (Table 1). She was given broad-spectrum antibiotics (blood cultures were eventually negative) and 3 daily pulses of intravenous solumedrol (1000 mg) with rapid defervescence. Nevertheless, the platelet count continued to decline, and her ferritin continued to rise, with worsening hypofibrinogenemia. Cyclosporine was increased to 300 mg daily (4.8 mg/kg, with serum levels in the therapeutic range), and she received 2 additional pulses of solumedrol, and intravenous immunoglobulin (IVIG; 1.5 gm/kg). On the fifth hospitalization day, anakinra was increased to 100 mg every 6 h (6.7 mg/kg/day) because of persistent thrombocytopenia (28,000/mm3); hyperferritinemia (5082 ng/ml); and an acute drop in fibrinogen to 30 mg/dl (Table 1). Every 6 h was chosen as the interval to increase benefit because anakinra has a short half-life (∼4 h) and a high therapeutic window1. Within 24 h, she had complete resolution of her malaise and her laboratory measures significantly improved (Figure 1): platelets 80,000/mm3; ferritin 2156 ng/ml; and fibrinogen 177 mg/dl (Table 1). Six days after high-dose anakinra, she was discharged with no signs of clinical disease activity and the following laboratory values: ferritin 1125 ng/ml; CRP 1.14 mg/dl; platelets 237,000/mm3; triglycerides 524 mg/dl; and fibrinogen 171 mg/dl (Table 1). Discharge medications included prednisone 40 mg twice daily, cyclosporine 300 mg daily, and anakinra 100 mg every 6 h. Prednisone and cyclosporine were tapered over 18 weeks, while she still took anakinra 4 times daily. Anakinra was discontinued 7 months after discharge, and the patient is taking no immunosuppressive therapy 2.5 years after discharge.
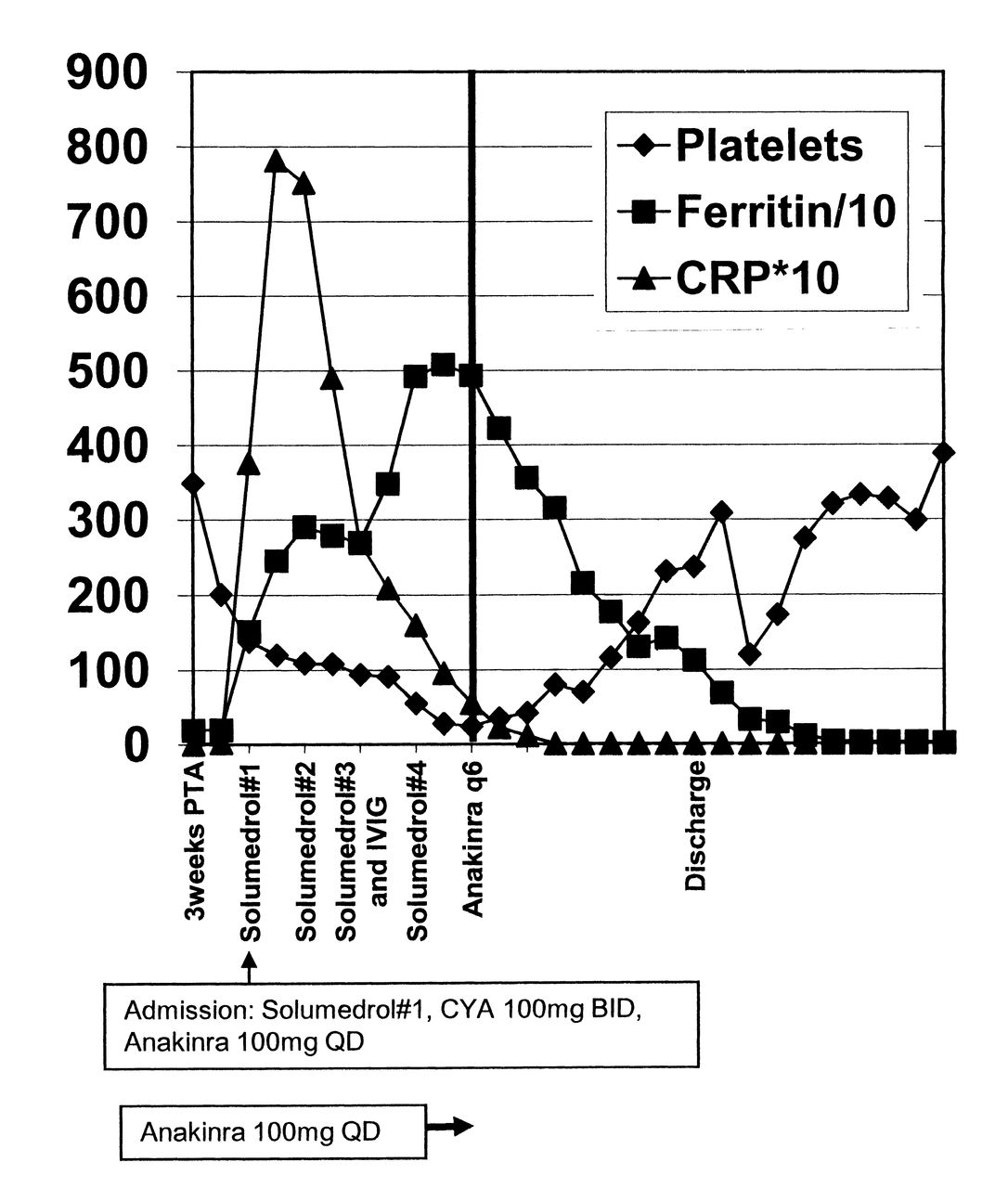
Response of macrophage activation syndrome measures to higher-dose anakinra. Platelet count (× 103/mm3), ferritin level (ng/ml divided by 10), and CRP level (mg/dl × 10) are graphed on the Y axis versus time in days (X axis). The vertical bold line represents increasing anakinra from 100 mg daily to 100 mg every 6 h (400 mg/day). Solumedrol (1 g intravenously), intravenous immunoglobulin (IVIG), and cyclosporine (CYA) administrations are noted. Hospital discharge is noted; PTA: prior to admission.
Laboratory data.
This case highlights the importance of aggressive inhibition of IL-1 with anakinra in children with sJIA complicated by refractory MAS. Up to 40% of children with sJIA develop subclinical MAS, including the diagnostic hallmark of hemophagocytosis on bone marrow biopsy2. Another 10% develop overt MAS, prompting aggressive antiinflammatory therapy2. Included among the reactive lymphohistiocytosis syndromes, MAS is a condition of cytokine storm, which may result in nonremitting high fever, encephalopathy, hepatitis, pancytopenia, coagulopathy, and death in up to 22% of patients3. In addition to treatment of ongoing hemodynamic instability, seizure, and bleeding, initiation of immunosuppressive therapy is urgently required for MAS, traditionally including various combinations of high-dose glucocorticoids, cyclosporine, etoposide, thalidomide, antithymocyte globlulin, and IVIG, which carry high risks of infection and other morbidities4.
Disease activity in sJIA is characterized by high levels of IL-1β, a prototypic proinflammatory cytokine5. IL-1 receptor antagonist (IL-1ra) is a naturally occurring IL-1 inhibitor normally produced by macrophages and other cells in response to IL-1, endotoxin, or other infectious pathogens. Anakinra is a recombinant, injectable human IL-1ra with a short half-life (∼4 h) that demonstrated remarkable efficacy in children with refractory sJIA5. Several case reports and series have reported successful use of anakinra in treating patients with MAS who were refractory to medical treatments including glucorticoids, etoposide, cyclosporine, methotrexate, IVIG, etanercept, and infliximab, with significant clinical response within days, including the chance to effectively taper glucocorticoids6,7,8,9,10,11.
In the setting of inflammatory diseases such as sJIA, greater concentrations of the naturally occurring IL-1ra may be required to antagonize the affects of IL-1 abundance, as in neonatal-onset multisystem inflammatory disease, a similar IL-1-mediated autoinflammatory disease in which patients may require doses as high as 10 mg/kg/day, with no apparent increase in infectious complications12. Although theoretic potential toxicities of newer therapeutics such as anakinra may result in appropriate caution, the well-known toxicity of traditional agents such as prednisone, cyclosporine, and etoposide are always concerning and include infection, growth retardation, osteoporosis, nephrotoxicity, neurotoxicity, and even death.
Anakinra, at higher than standard dosing, appears to be a novel, effective, and quick-acting agent. Earlier initiation of anakinra in patients with potentially life-threatening MAS may better treat the underlying mechanisms mediating disease while minimizing toxicity of other more nonspecific agents. One may consider use of higher-dose anakinra in patients with medically refractory MAS. Further prospective study of this approach is warranted.
Footnotes
-
Dr. Cron serves as a consultant to Novartis regarding canakinumab and to Genentech regarding tocilizumab therapy for systemic juvenile idiopathic arthritis.








{kind=link}