

Abstract
Objective. Systemic juvenile idiopathic arthritis (SJIA) frequently leads to disability and damage. Predictive factors for a poor outcome include persistent systemic features and younger age at onset. We describe and analyze disease features in patients with early-onset (EO) SJIA (disease onset before age 18 mo) and compare them to patients with later-onset (LO) disease.
Methods. Clinical features at onset, activity measures (occurrence of macrophage activation syndrome, remission), and outcome measures for disability [Childhood Health Assessment Questionnaire (CHAQ) ≥ 0.5] and damage [radiographic joint destruction, Juvenile Arthritis Damage Index (JADI) score, growth retardation] observed during followup were analyzed retrospectively in patients with SJIA followed for ≥ 3 years since disease onset.
Results. In total 132 patients were included. SJIA started at age ≤ 18 months in 19 (14%) patients and at a later age in 113 (86%) children. At onset, serositis (p < 0.01) and hepatomegaly (p < 0.05) were more frequent in EO patients, who also exhibited lower hemoglobin levels (p < 0.03) and higher platelet counts (p < 0.03) than patients with LO. Macrophage activation syndrome occurred in 20 patients (11 EO and 9 LO; p < 0.0001). Remission was achieved by 49 patients (37%; 4 EO and 45 LO). At last visit, destructive hip disease (p < 0.04), growth retardation (p < 0.01), radiographic damage (p < 0.02), and disability (p < 0.04) were more frequent in patients with EO disease, who had higher JADI scores (p < 0.003).
Conclusion. Patients with EO exhibited a more aggressive and destructive disease course than patients with LO SJIA.
Systemic juvenile idiopathic arthritis (SJIA) is one of the most severe forms of JIA, frequently leading to severe disability and significant mortality1. Its clinical course is variable in severity and periodicity and frequently leads to significant damage and disability2. Prognostic factors for an unfavorable outcome include persistent systemic features and younger age at onset3,4.
Age at disease onset is variable in SJIA, with a median of 6 years. In a meaningful proportion of children disease onset occurs at an early age5, exceptionally in infants6. Certain physiologic developmental processes, such as patency of transphyseal vessels in the femoral head7 and maturation of the immune system8, normally wane during the second year of life, by age 18 months. We speculated that the clinical expression of SJIA starting before age 18 months may be different from the clinical picture exhibited by patients in whom the disease has a later onset.
We reviewed our experience in patients with SJIA to assess the clinical manifestations and outcomes in children with disease onset occurring before age 18 months and to compare them to those of patients with a later disease onset.
MATERIALS AND METHODS
We retrospectively reviewed the records of patients with a diagnosis of SJIA according to the criteria of the International League of Associations for Rheumatology9. The patients attended the rheumatology clinic of the Service of Immunology and Rheumatology of the Hospital de Pediatría Garrahan, Buenos Aires (a tertiary pediatric referral facility), between January 1995 and December 2010. Criteria for inclusion in the study comprised complete records and continuous followup ≥ 3 years since disease onset (defined as the first 6 months of the disease course). Patients were followed periodically and data were prospectively collected and recorded in a database. Variables recorded were sex, age, delay in diagnosis (time elapsed between first symptom compatible with SJIA and the formulation of a correct diagnosis), highest observed number of active joints, presence of polyarthritis (≥ 4 affected joints), site and pattern of joint involvement (axial vs peripheral, small vs large, upper limbs vs lower limbs), presence of rash, serositis, generalized lymphadenopathy, splenomegaly, hepatomegaly, highest recorded white blood cell count, platelet count, and erythrocyte sedimentation rate, and lowest recorded hemoglobin level at disease onset; pattern of disease course (monocyclic, intermittent, or persistent) according to Lomater, et al10, site and pattern of joint involvement, immunomodulatory therapy [corticosteroids, methotrexate (MTX), biologics], and death during the disease course; occurrence of macrophage activation syndrome (MAS; diagnosed by the treating physician on the basis of a combination of fever, cytopenias, liver dysfunction, and coagulopathy, irrespective of the presence of hemophagocytosis in the bone marrow or other organ biopsy), presence of growth failure [defined as lower than the third percentile of height for age and crossing at least 2 centiles (5%, 10%, 25%, 50%, 75%, 95%) on growth chart], disability [defined as Childhood Health Assessment Questionnaire (CHAQ), Argentinian version11, score > 0.5], radiographic damage (defined as presence of bony erosions/fusion), destructive hip disease (erosions, collapse, protrusio acetabuli, avascular necrosis, or fusion), and remission according to Wallace, et al12 during disease course and at last observation. Remission criteria were applied retrospectively to visits recorded before 2004. Clinical damage measured by the Juvenile Arthritis Damage Index (JADI)13 was scored at the last visit. The JADI score defines the presence of persistent (≥ 6 months) articular and extraarticular damage. Articular damage assessment relies on the presence of limitation of range of motion, contractures, and other joint deformities that are not related to joint inflammation, and/or presence of severe bone involvement. Extraarticular damage includes 13 items in 5 different systems, which are scored as either 0 or 1 according to whether damage is absent or present. The maximum total JADI score is 89 (72 for the articular and 17 for the extraarticular components). JADI was scored only in patients who were last observed in our clinic in the period 2006–2010.
All patients were treated according to the local practice guidelines. The usual practice during the observation period was to administer MTX to patients who did not experience improvement during the first month of nonsteroidal antiinflammatory drugs plus systemic corticosteroid therapy. Use of biologic agents was reserved to patients who showed persistent activity despite the use of parenteral MTX for 3 months. Biologic agents were first prescribed in December 1999. Radiographic assessment includes plain radiographs of all symptomatic joints repeated every 1 to 2 years.
Patients were divided into 2 groups: early onset (EO; before 18 months) and late onset (LO; after 18 months). Patients with EO or LO were compared using chi-square (or Fisher exact) and Mann-Whitney U tests. Kaplan-Meier survival analysis was used to estimate the proportion of patients who achieved remission during the disease course (comparison between groups was performed with the log-rank test). A p value < 0.05 was considered significant. Statistix 9 (Analytical Software) was used for all analyses.
RESULTS
Of the 192 patients with SJIA who attended our clinic, 132 met the inclusion criteria and were eligible for the study. A total of 60 patients (4 EO, 56 LO) were excluded because they had a followup shorter than 3 years (n = 36) or the records were not informative about clinical features at onset or during the disease course (n = 24).
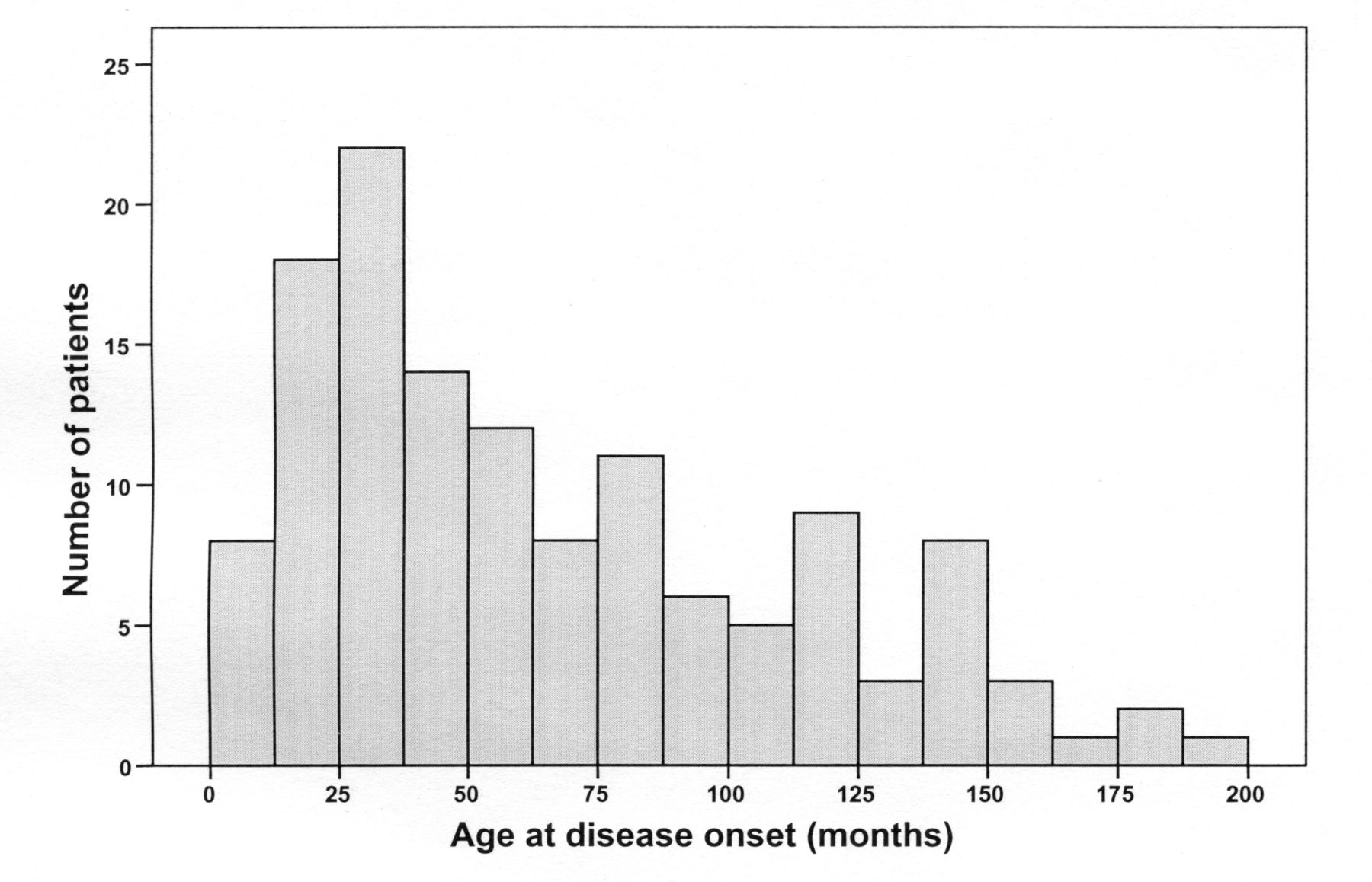
There was a female predominance (1.4:1); median age at disease onset was 60 months (Figure 1). There were 19 patients (14%) in the EO group and 113 (86%) in the LO group. Seven patients with EO (37%) and 56 (50%) with LO had disease onset and received their first specialized care before December 1999.
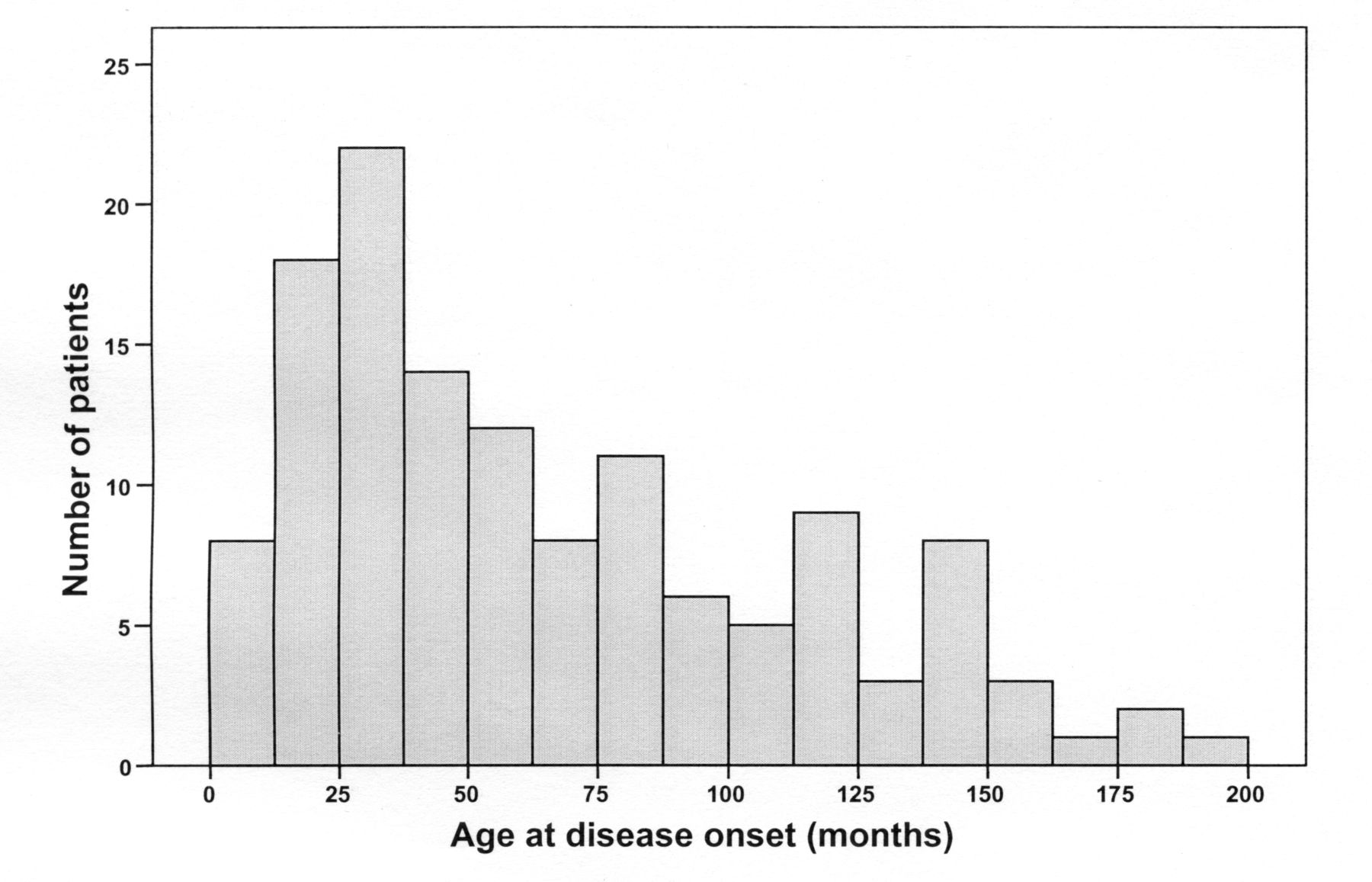
Age at disease onset in a cohort of patients with systemic juvenile idiopathic arthritis.
Table 1 summarizes patient demographics and clinical features at onset. Seven children with EO (37%) exhibited very low hemoglobin levels (< 80 g/l), while such levels were recorded in 14 patients (12%) with LO at disease onset (p < 0.007). Clinical features at onset in patients with a followup < 3 years (median 1.0 yr) were not significantly different from those of patients with longer followup. Small joint and upper limb involvement was more frequent in the EO group at onset, but there were no differences in joint involvement patterns between the 2 groups during the disease course (Table 2).
Demographic and clinical features at onset in 132 patients with systemic juvenile idiopathic arthritis. Data are median (interquartile range) unless indicated otherwise.
Patterns of joint involvement at onset and during followup in 132 patients with systemic juvenile idiopathic arthritis. Values are number (percentage) of patients.
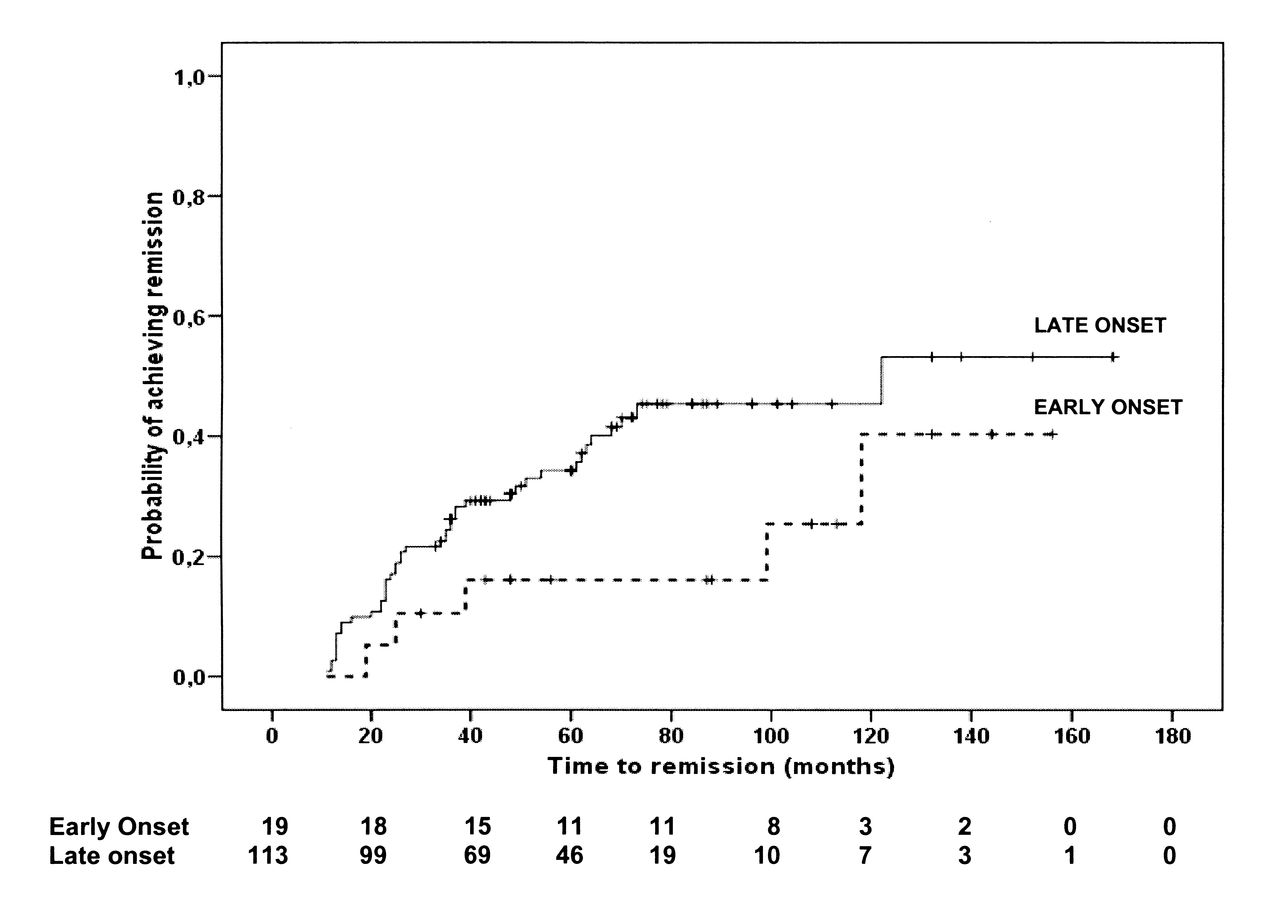
Immunomodulatory drugs used for treatment of SJIA in this cohort included corticosteroids, MTX, and biologic agents (Table 3). Median time between disease onset and start of treatment with biologics was 33 months (53 months in the EO group, 31.5 months in the LO group; p = 0.34). Use of antitumor necrosis factor (TNF) agents was followed by remission in 11 patients (22%; 1 EO and 10 LO). Overall, remission was eventually achieved by 49 patients (37%) in the EO (n = 4) and the LO (n = 45) groups. The cumulative incidences of achievement of remission are shown in Figure 2. There were no significant differences between the 2 groups. Remission off-therapy was observed in 12 children (1 EO and 11 LO). During the disease course, MAS occurred in patients with EO more frequently than in patients with LO (p < 0.0001). Of note, 6/11 MAS episodes recorded in patients with EO occurred as the first manifestation of SJIA, while MAS was the presenting symptom in 3/9 patients with LO.
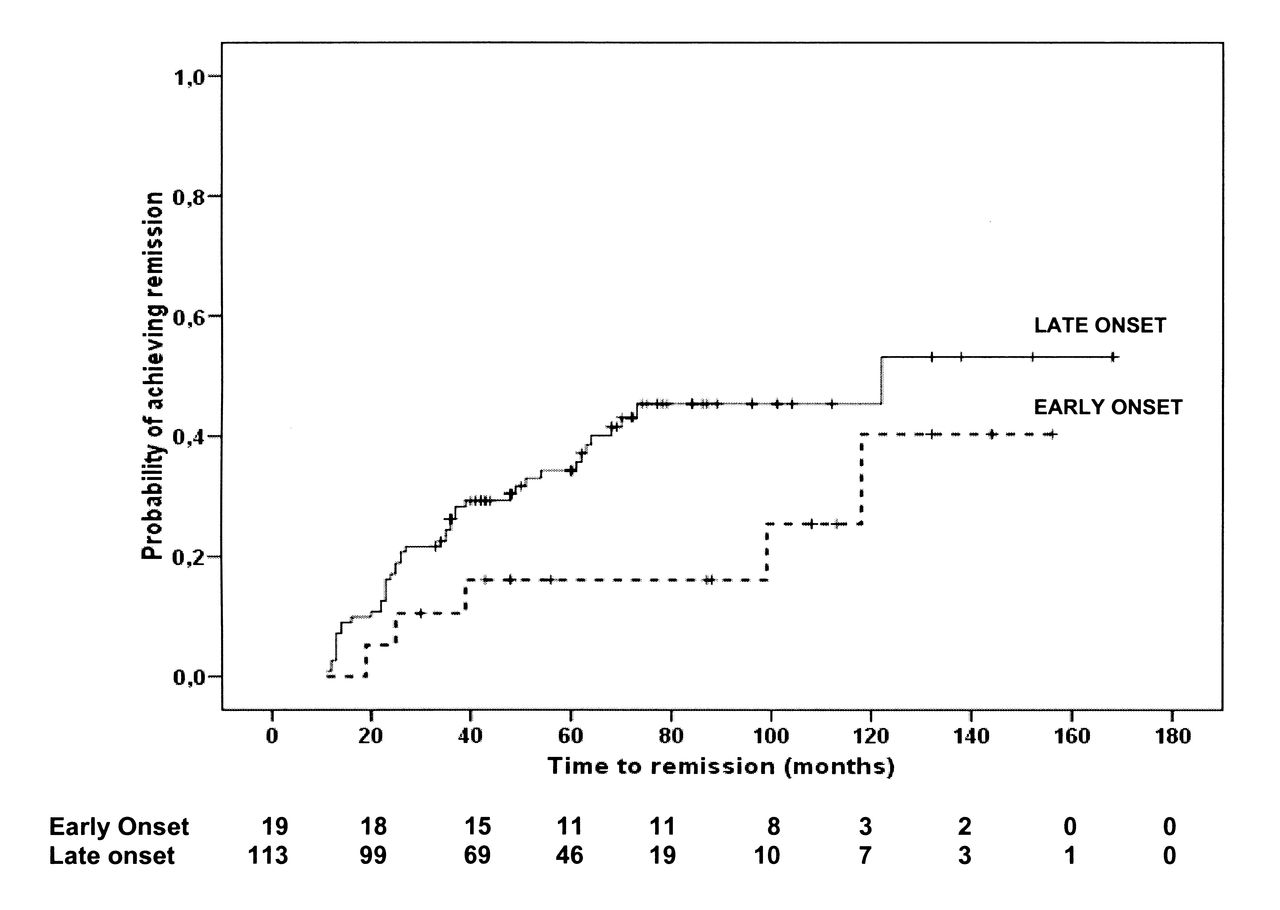
Kaplan-Meier estimates for achieving remission in patients with early-onset or later-onset systemic juvenile idiopathic arthritis. Number of patients at risk is shown for each group at several timepoints.
Disease course pattern and therapeutic strategies used in 132 patients with SJIA. Values are number of patients (percentage) or median (interquartile range).
At last observation, destructive hip disease, radiographic damage, and disability were more frequent in patients with EO (Table 4). JADI (global) was scored in 75 patients (16 EO, 59 LO): it was significantly higher in the patients with EO. Two patients in the LO group died during the observation period (multiorgan failure due to MAS, and sepsis, respectively).
Clinical features and outcomes observed at 3 years after disease onset and at last visit in 132 patients with systemic juvenile idiopathic arthritis. Values are number of patients (percentage) or median [interquartile range (IQR)].
DISCUSSION
SJIA is a heterogeneous disease. Some patients have very benign and monophasic forms, while others have devastating, progressive disease with very poor outcomes10. Predictive factors for poor outcome have been identified by several investigators. Male sex, corticosteroid therapy, persistent systemic symptoms, cervical involvement, and radiographic progression during the first year predicted a chronic, polyarticular course, articular and extraarticular damage, and disability in different small cohorts of patients with SJIA3,14,15,16,17,18,19. Modesto, et al4 demonstrated that generalized lymphadenopathy and age at onset < 8 years as well as hip involvement and polyarthritis at 6 months after start of the disease were associated with a bad outcome in a cohort of 91 patients with SJIA. On the other hand, Oen, et al20 found an association between younger age at onset and shorter active disease duration in their 16 patients with SJIA.
SJIA patients with very early onset (before age 18 months) have a distinct disease course according to our findings in this large single-center sample. They show more pronounced systemic inflammatory features at disease onset, more frequent development of MAS, and they exhibit poorer functional and structural outcomes than patients with later-onset disease. The reasons for this particular course are not well understood. A combination of genetic, anatomic, and physiological factors might be involved in the clinical expression of SJIA at different ages5. Hypothetically, immature joints may be more susceptible to inflammation-driven damage in patients with younger age at onset. Earlier hip inflammation and damage accrual are likely to cause delayed and abnormal standing and walking, as well as permanent disability.
The rationale for classifying the EO group as patients with disease onset before age 18 months has a physiologic background. We speculated that children with disease onset before 18 months may be exposed to particular anatomical and immunologic conditions that may originate particular clinical features and outcome. Certain physiologic developmental processes, such as maturation of the immune system and patency of transphyseal blood vessels, normally occur in the first months of life and reach an end at about 18 months7,8. At a younger age the immune system depends primarily on the innate immunity, whereas in older children the adaptive system has been well developed. SJIA, a predominantly innate immune-mediated disease21, may exhibit certain characteristics in a very young child, in whom there is predominant innate immunity, that are different from those in an older patient. The striking occurrence of MAS observed in the first months of the disease course in our patients with very early-onset SJIA seems to support this idea. Hypothetically, the patent transphyseal vessels may facilitate the access of inflammatory mediators to the epiphyses and joints (similarly to the hematogenous spread of microorganisms to the joints in infant septic arthritis). It is noteworthy, however, that in the first months of the disease, hip arthritis was observed in a small proportion of patients with EO SJIA. Eighty percent of patients with EO SJIA who exhibited evidence of hip involvement during disease course eventually developed destructive hip disease, demonstrating the severity and possibly the persistence of longstanding inflammation in this joint. Early hip involvement was identified as a predictor for bad prognosis by Modesto, et al4 in a cohort of SJIA patients with an overall age at onset comparable to that of our patients. In that study, the authors found a percentage of patients with early hip involvement similar to that we observed, 27% and 37%, respectively. There is a possibility that hip arthritis was undetected through clinical examination during disease onset in some young patients in our study. We did not perform ultrasound or magnetic resonance imaging (MRI) examinations to detect hip involvement on a routine basis in our patients. Hip ultrasound and MRI can reveal synovial inflammation in hips that is not detected by clinical examination in patients with JIA22,23.
There were no significant differences between the 2 groups in immunomodulatory therapeutic strategies for SJIA, except for the use of biologics. These agents were used in a higher proportion of patients with EO, possibly reflecting the more severe disease in this group. There was a significant delay between disease onset and initiation of a biologic therapy in both groups. Reasons for this delay are various: a proportion of patients had 1 or more periods of disease inactivity/improvement alternating with periods of activity/worsening before the decision was made to add a biologic agent; administrative issues may have caused additional delay; and nearly 40% of patients had onset of their disease (and refractoriness to MTX in some cases) before any biologic agent was available, but were eventually treated with these agents when they became available. However, none of these factors seems to have differed significantly between the 2 groups. Although not statistically meaningful, a longer elapsed time between disease onset and start of biologic therapy in the EO group may have had an effect on disease outcome. TNF inhibitors, the biologic agents used most commonly in our cohort, have demonstrated variable efficacy in the treatment of patients with SJIA, and their use may induce remission in a proportion of children, as seen in the present and previous studies24. It is expected that the course of SJIA will be affected by the growing numbers of patients treated more promptly with new, more effective biologic strategies such as interleukin 6 (IL-6) and IL-1-inhibiting agents25,26.
Our study should be interpreted in light of its limitations, such as retrospective retrieval of data and assessment of patients from a single center. The small sample and the asymmetric size of groups precluded multivariate analysis of the data and challenged statistical reliability. There is a possibility of associations found in this cohort changing because of the addition of more cases or the extension of followup and changing outcome variables. Moreover, referral and selection bias cannot be excluded. The limitations of the clinical examination, as addressed, may have imposed an error in the detection and recording of hip and other joint involvement. The lower hemoglobin levels exhibited by younger-onset patients during disease onset may be caused at least partially by physiological and nutritional factors. The excluded patients who had an insufficient followup time may constitute another source of bias. Because their last visit occurred a median of 12 months after disease onset, no comparisons could be made about the pattern of disease course or features present at last visit. However, these patients did not exhibit any significant clinical differences at onset. Lastly, CHAQ score is probably not as representative of true functional capacity in younger children as it is in older children because it depends upon proxy reporting by the parent. Parents probably score their child’s functional ability as greater than the child would score himself or herself if he or she could11.
Patients with SJIA starting before age 18 months show more systemic inflammatory features and a poorer outcome than children with later disease onset. This observation, which needs confirmation in larger populations in multicenter studies, should prompt rheumatologists to initiate early aggressive therapy and close followup in this group.
- Accepted for publication November 23, 2012.








{kind=link}
{kind=link}