
Abstract
Objective. The TRAF1 genetic region conferring susceptibility to rheumatoid arthritis (RA) has been reported to associate with radiological damage. We aimed to test RA genetic susceptibility markers for association with a continuous measure of radiological damage over time using longitudinal modeling techniques.
Methods. Sixty-seven RA susceptibility variants were genotyped in 474 patients in the Early Rheumatoid Arthritis Study (ERAS) using Sequenom MassArray technology. Correlation between genetic markers and Larsen score was assessed longitudinally using zero-inflated negative binomial regression to include repeat measurements in the same individual at different timepoints. Genetic markers associated with radiological damage in ERAS were tested using the same modeling techniques on previously published data from the Norfolk Arthritis Register (NOAR).
Results. The single marker associated longitudinally with Larsen score in ERAS (p = 0.02) and in NOAR (p = 0.04) was rs2900180 at the TRAF1 locus. Analysis of individual timepoints in ERAS showed that rs2900180 displays its effect primarily on the extent of Larsen score early in the disease course. Combined longitudinal analysis of the 2 cohorts suggests further association of several loci with Larsen score (KIF5A, PTPN22, AFF3, TAGAP) and therefore a significant accumulation of RA severity markers among RA susceptibility markers (p = 0.016).
Conclusion. The marker rs2900180 is associated with the extent of radiological damage in the ERAS cohort. This represents the second independent study correlating rs2900180 at the TRAF1 locus with radiological severity in RA. Replication in a large dataset is required to establish the role of other RA susceptibility loci in disease severity.
- GENETIC ASSOCIATION
- RADIOLOGICAL DAMAGE
- EROSIONS
- LONGITUDINAL MODELING
- RHEUMATOID ARTHRITIS
- TRAF1
Rheumatoid arthritis (RA) susceptibility variants within the genomic interval flanked by the tumor necrosis factor (TNF) receptor-associated factor-1 and complement component 5 genes (TRAF1/C5) were first described in 2 independent disease association studies in 20071,2. The studies reported association with 2 different markers in this region: rs2900180 and rs3761847. The markers rs2900180 and rs3761847 are in moderate linkage disequilibrium (LD; r2 = 0.69) and it is not clear which is more closely associated2. Following the discovery of this locus, the association with RA susceptibility was confirmed in independent cohorts3 and by metaanalysis4,5,6 and is more strongly associated with anticitrullinated peptide antibody (ACPA)-positive RA7,8.
The TRAF1/C5 locus variants have since been investigated for correlation with RA outcome. No association has been detected with continuation on methotrexate as a monotherapy9, response to TNF blockade therapy10, or premature mortality11,12. However, the markers rs10818488 (r2 = 0.97 with rs3761847) and rs2900180 have both been correlated with radiological severity1,13. Although the effects at this locus are modest, detection of a relationship between radiological outcome and the TRAF1 locus in 2 independent studies is intriguing and warrants further investigation in independent cohorts. A recent study replicated the association of rs3761847 with radiological damage in Iceland, but could not replicate this association or the association of rs10818488 in 6 other cohorts originating from different countries14. Another recent study showed rs10818488 to be associated with radiological damage in Egyptian patients15. However, there have been no published reports investigating rs2900180 in an independent population. Importantly, the various studies above differ in study design, populations, or statistical approach, and most of them are underpowered. Moreover, the definition of radiological damage across studies is heterogeneous. Measures of radiological outcome have been presence of bone erosions and extent of a continuous Larsen or Sharp-van der Heijde score, either cross-sectionally or its progression over time16.
There are now > 60 single-nucleotide polymorphism (SNP) markers mapping to over 30 loci with confirmed or highly suggestive evidence for association with RA susceptibility1,2,5,6,17,18,19,20. Although several studies have investigated non-HLA RA susceptibility markers as putative markers of disease outcome, no convincing genetic marker of erosive disease has yet been identified outside the HLA16. In our study, we aimed to test the hypothesis that RA susceptibility markers also modulate disease severity. First, we investigated 67 genetic predictors of RA development, including rs2900180 and rs10760130 (r2 = 1 with rs10818488), as potential determinants of radiological outcome using longitudinal statistical modeling to increase power. Markers with suggestive evidence of association in the Early Rheumatoid Arthritis Study (ERAS) were analyzed again in previously published data from the Norfolk Arthritis Register (NOAR) using the modeling techniques described here. Longitudinal modeling has the advantage of incorporating all the information on radiological outcome assessed at different timepoints into the analysis and can incorporate multiple measures for the same individual. Second, we combined these data to further increase detection power.
MATERIALS AND METHODS
Patients
ERAS recruited adult patients with RA between 1986 and 1998 from 9 UK centers as described21. Briefly, entry criteria included < 2 years of symptoms and no prior treatment with disease-modifying antirheumatic drugs. Clinical and demographic data, collected at baseline assessment, included sex, age at symptom onset, and disease duration at registration. Hand and feet radiographs were taken systematically at baseline, 6 months, and 1 year after registration and yearly thereafter, for 15 years. Radiographs at baseline and yearly thereafter up to Year 9 were scored using the Larsen method22. Erosive disease was defined by the presence of at least 1 joint with a Larsen score ≥ 2 (interruption of Corticalis continuity). Radiographs were scored by a single medically qualified researcher (CS) with built-in observer reliability/repeatability mechanisms (intrareader correlation coefficient 0.97), as described23. A blood sample was collected at recruitment to permit genetic studies. Only whites in the UK were included in this analysis. ERAS received ethical approval from the West Hertfordshire Local Research Ethics Committee and subsequently from the Caldicott Guardian. Informed consent was obtained from all patients.
NOAR is a primary care-based inception cohort of white UK patients with recent-onset inflammatory polyarthritis (IP) followed prospectively from disease onset to almost 20 years. Details of the NOAR are described elsewhere13,24. Briefly, adults aged ≥ 16 years with swelling of 2 or more joints lasting 4 weeks or longer were referred to NOAR. Radiographs of the hands and feet were performed at baseline, Year 1, and Year 2 if required to establish the diagnosis of RA, and systematically at Year 5 for all patients and scored using the Larsen technique. Radiographs were read independently by 2 observers who were blinded to the sequence. In preliminary studies, the intraobserver and interobserver agreements for the presence of erosions were 90% and 81%, respectively25. Disagreement on the erosion status was settled by arbitration by a third investigator. Consensus-reading sessions were held to settle minor disagreements on scores. Although no formal interobserver correlation between ERAS and NOAR was performed, the high within-study correlations make a high inter-study correlation likely. The NOAR cohort shares similar characteristics with the ERAS cohort. Ethical approval for the study was obtained (MREC 99/8/84) and informed consent was signed by all patients.
The goal was to identify/replicate genetic markers of future disease severity to guide treatment decision early at disease onset, before American College of Rheumatology criteria for RA were satisfied. The power of the study and the discriminatory capacity of a future genetic test are larger early in the disease course than later; we therefore performed the analysis on IP in NOAR.
Genotyping
SNP markers were genotyped in ERAS patients using Sequenom MassArray technology, in accord with the manufacturer’s instructions. Markers were selected from large case-control disease association studies in RA or resultant metaanalyses1,2,5,6,17,18,19,20. Patients with > 10% missing genotype data and SNP with < 90% genotyping success rate were excluded prior to analysis as part of quality control procedures. HLA-DRB1 “shared epitope” alleles (HLA-SE: coded as 0, 1, or 2 copies) were detected using sequence oligonucleotide typing, as described26. The following HLA-DRB1 genotypes were considered as SE alleles in ERAS or in NOAR: 0101, 0102, 0104, 0105, 0107, 0108, 0110, 0111, 0401, 0404, 0405, 0408, 0409, 0410, 0413, 0416, 0419, 0421, 0423, 0426, 0428, 0429, 0430, 0433, 0434, 0435, 0438, 0440, 0442, 0443, 0445, 1001, 1113, 1126, 1134, 1402, 1409, 1413, 1417, 1419, 1420, 1421, 1429, 1430, 1431, 1432, 1434, 1441, and 1446, 1447, 1448. The genotyping of SNP markers for the NOAR cohort has been published using cross-sectional multivariate analysis13. All r2 values mentioned in this work to qualify the extent of LD between markers have been calculated using the HapMap CEU reference panel.
Longitudinal multivariate analysis
To incorporate multiple records per patient over time into the same model, Larsen score was considered as a time-dependent longitudinal trait. The Larsen score, in particular at early followup, is not normally distributed, with an excess of zero values (nonerosive patients). Consequently, a zero-inflated negative binomial (ZINB) regression model27 was fitted for every SNP to assess association with Larsen score. This regression model has 2 components: one for the probability that the score is non-zero (susceptibility model), and a separate model for the magnitude of the score, given that it is non-zero (severity model). A given predictor may contribute to either of these models, or both. To allow for the within-subject correlation, a clustered Huber/White/sandwich estimate of the SE of the model measurements was used28.
Disease duration at the time of Larsen score measurement was defined as disease duration at registration plus number of years of followup. To incorporate timepoints up to the 5-year followup for all patients, the analysis was performed for disease duration < 7.5 years. To account for change in Larsen score over time, disease duration was included in the regression model. Both linear and quadratic terms in disease duration were included, to allow for nonlinearity in the changes in Larsen score with time. The ZINB regression provides 2 separate point estimates and p values for every SNP, for the susceptibility and severity submodels. The 2 p values were combined and reported as ZINB combined p value. This was not possible for the point estimates, because they have different units. Instead, the marginal effect dy/dx of the model for the average value of x (i.e., the SNP) was calculated and reported as “time averaged effect size” in Larsen units. The modeling was undertaken in each cohort separately and then a combined analysis of both cohorts was performed by pooling observations from the individual cohorts into a unique dataset and adjusting for cohort of origin by fitting terms for the interaction between cohort and disease duration. This accounts for the difference in the velocity of disease progression between the 2 cohorts.
This approach allowed incorporation of all radiographs taken at different timepoints for all patients in every cohort into a single model, and did not require that only timepoints are taken into account, where radiographs have been performed systematically for all patients in a cohort.
Cross-sectional multivariate analysis
Correlation between genetic markers and Larsen score was assessed cross-sectionally at baseline and at subsequent followups with a ZINB model if erosive and nonerosive patients were included in the analysis, or with negative binomial regression if only erosive patients were considered. Logistic regression was used to assess the effect of an SNP on the probability of a subject having erosions. Based on previous experience and because of low counts of homozygotes for the minor allele for most SNP, we assumed, as for the above-mentioned longitudinal analysis, a dominant model for all analyses, i.e., carriage of at least 1 copy of the minor allele versus no copy12. Adjustment in cross-sectional analyses was made in every model for symptom duration at baseline.
All analyses were performed using Stata version 10.1 software (StataCorp. 2007, Stata Statistical Software Release 10, StataCorp LP).
Multiple testing
No correction for multiple testing was applied for the p values presented in the results tables. Several tested SNP are in LD and map to the same genetic locus (Appendix 1). The number of independent tests is therefore smaller than 67. There is, however, no consensus on the LD threshold to use for multiple testing corrections. Moreover, under a prior hypothesis of no association, a more stringent correction for multiple testing should be applied than in replication studies. Prior evidence of association, as is the case for several markers including TRAF1/C5 markers, does not formally require corrections for multiple testing.
RESULTS
ERAS cohort
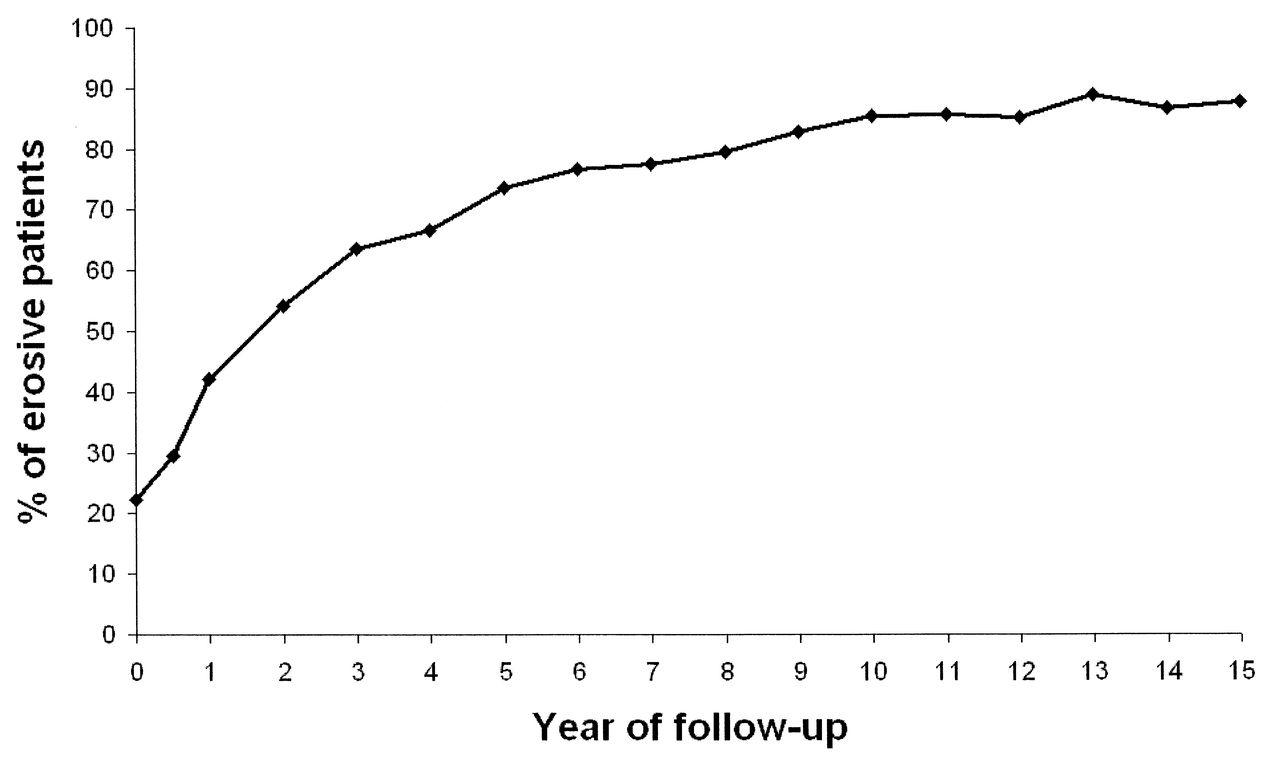
Within the first 5 years of followup, nearly three-quarters of the patients in the ERAS cohort developed bone erosions (Figure 1). To model radiological damage as a continuous trait over the years when the vast majority of patients develop erosive disease, correlation between SNP markers and the Larsen score included all timepoints up to a disease duration of 7.5 years. Sixty-seven SNP (Appendix 1) were genotyped in 474 DNA samples from the ERAS cohort. One SNP (rs934734) failed to genotype. Therefore, following quality control, 66 SNP remained available for analysis in 445 samples (94% sample success). Summary cohort characteristics are presented in Table 1.
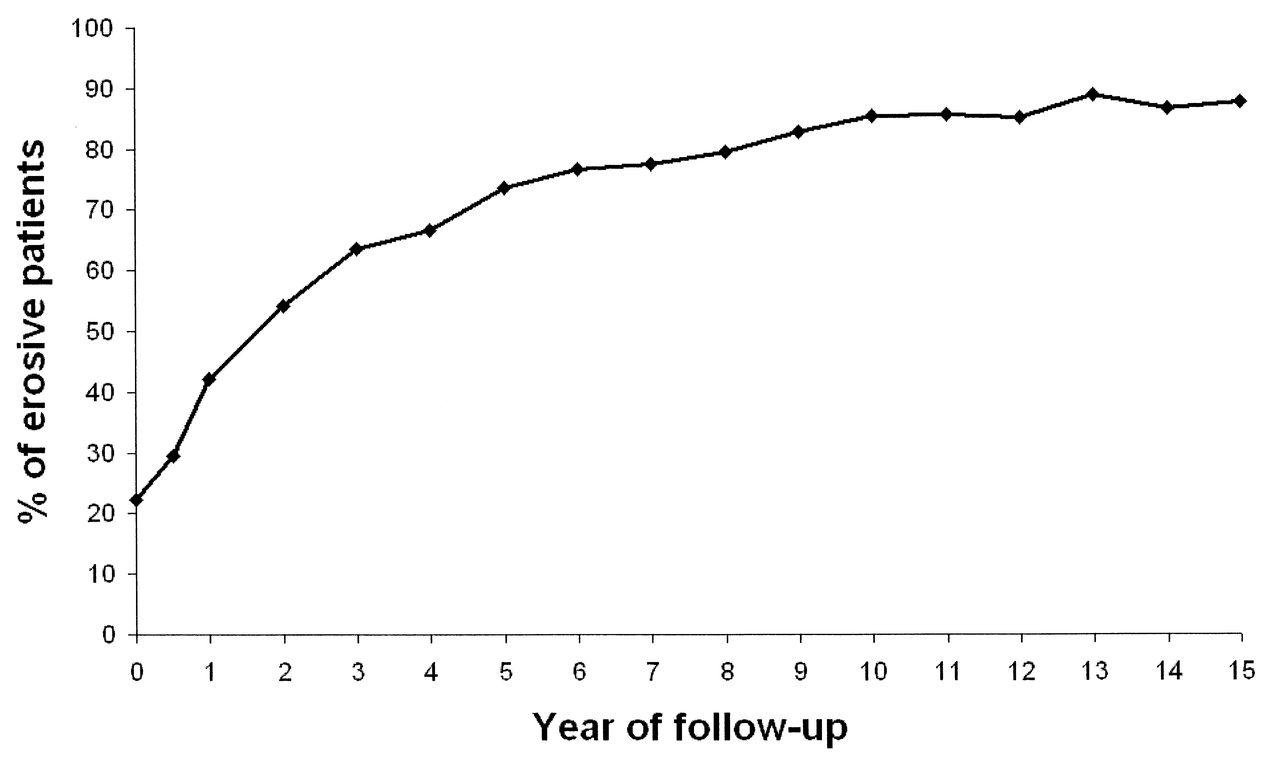
Percentage of erosive patients at every followup in the Early Rheumatoid Arthritis Study.
Summary cohort characteristics.
Longitudinal analysis of radiological outcome in ERAS
There were on average 5.4 Larsen scores (SD 1.7, range 1–8) per patient in ERAS when analysis was limited to a disease duration of 7.5 years. Larsen scores at every timepoint were highly correlated with scores at the previous timepoint, especially later in the disease course, when radiological progression slows down (average Spearman correlation coefficient between subsequent measurements: 0.87). Taking this into account, and assuming a minor allele frequency (MAF) of around 40% (i.e., TRAF1/C5 markers or SE), the power to detect a time-averaged difference of 2 Larsen units at a significance level of 0.05 between carriers of the minor allele and non-carriers is 34%, using a longitudinal approach. The power rises to 64% for a difference of 3 Larsen units and to 97% for 5 units. The estimated sample size required to detect differences in Larsen scores of 2 units is about 2000 patients, assuming 5 repeated measurements correlated at 0.87 and a MAF around 10% (i.e., PTPN22). The shared epitope showed a highly significant association with Larsen score (ZINB combined p value: 5.12E-05) with a strong time-averaged effect size (marginal effect dy/dx): the carriage of at least 1 copy of the shared epitope increased the Larsen score on average by 5.36 Larsen units over the time period considered for analysis (Table 2 and Figure 2). Notably, the shared epitope is already significantly associated with Larsen score at baseline. Results for all tested SNP are in Appendix 1. Three SNP [rs2476601 (PTPN22), rs394581 (TAGAP), and rs2900180 (TRAF1)] displayed a ZINB combined p value < 0.05 for association with Larsen score (Table 2). Those markers were tested using the same modeling technique applied on a previously reported dataset in NOAR to serve as replication.

{kind=link}
{kind=link}
Predicted Larsen score as a function of disease duration and carriage of at least 1 copy of the shared epitope (left panel) or the minor allele of rs2900180 (right panel) in the Early Rheumatoid Arthritis Study. A zero-inflated negative binomial regression model is used, containing disease duration and the shared epitope (left panel) or the minor allele of rs2900180 (right panel) as independent variables. Predicted values for carriers of the shared epitope (left panel) or the minor allele of rs2900180 (right panel) are shown in black and for noncarriers in gray.
Regression analyses of correlation between genetic markers of rheumatoid arthritis (RA) susceptibility single-nucleotide polymorphisms (SNP) and Larsen score as a continuous time-dependent trait. Results for the 3 SNP displaying a zero-inflated negative binomial (ZINB) combined p value < 0.05 in the Early Rheumatoid Arthritis Study (ERAS) are shown together with the shared epitope and the TRAF1/C5 marker rs10760130. Results for those 5 markers using the same modeling technique applied on a previously reported dataset in the Norfolk Arthritis Register (NOAR) are presented to serve as replication. Results for all RA susceptibility SNP are available in Appendix 1.
The predicted values of the Larsen score for carriers versus noncarriers of the minor allele of rs2900180 in ERAS are plotted over time in Figure 2. Unlike the shared epitope, rs2900180 does not significantly affect radiological severity at baseline, but it does influence radiological progression, although the effect size is lower (time-averaged effect size dy/dx: 2.96). Adjustment for the shared epitope does not meaningfully affect the results (data not shown), and neither does adjustment for ever-positivity of rheumatoid factor (ZINB combined p value: 0.023; dy/dx: 2.98).
Longitudinal analysis of radiological outcome in NOAR
Detailed cohort characteristics and genotyping have been described13; basic cohort characteristics are presented in Table 1 and compared with the characteristics of the ERAS cohort. Both cohorts share many features; however, NOAR recruits patients with IP, while ERAS recruits only patients with a diagnosis of RA. Therefore, NOAR patients display a lower disease severity for the same disease duration. This does not represent a limitation, because the modeling technique described here accounts for disease duration and for the difference in kinetics between the 2 cohorts, when pooled.
Most (including rs2900180 and rs10760130) but not all of the RA susceptibility markers genotyped in ERAS have been reported in NOAR13, but analyzed using cross-sectional techniques (Appendix 1). However, the power to detect a difference of 2 Larsen units at the 0.05 significance level in NOAR at Year 5 for a MAF around 40% is only 40% when analyzed using a cross-sectional approach. Using a longitudinal approach increases the power to 83% in NOAR, although there were on average 1.5 Larsen scores per patient in NOAR (SD 0.7, range 1–4). This increase of power is mainly achieved by recruiting a significant proportion of patients who did not complete the 5-year followup period or had missing data at 5 years, but had a radiograph taken previously.
Therefore, the NOAR dataset was reanalyzed using longitudinal ZINB modeling including all timepoints up to a disease duration of 7.5 years, to serve as replication for the putative markers of radiological damage identified in ERAS. Only rs2900180 (TRAF1) showed an association with Larsen score in both cohorts (Table 2 and Appendix 1). Adjustment for the shared epitope in NOAR did not meaningfully affect the results (data not shown).
Cross-sectional analysis of TRAF1 markers of radiological outcome
Cross-sectional analyses of association between TRAF1 locus variants and Larsen score or the presence of erosions were performed at baseline and subsequent followups in ERAS and NOAR. The strongest associations, both with Larsen score and/or erosions, were found early in the disease course at timepoints when roughly half the patients displayed erosive disease. Because of differences in the overall disease severity in ERAS and NOAR, these timepoints are Years 2–3 in ERAS and Year 5 in NOAR (Table 3).
Cross-sectional analyses of association between TRAF1/C5 locus variants and Larsen score or the presence of erosions using a zero-inflated negative binomial regression (ZINB) on all patients, a negative binomial regression (NBREG) on erosive patients, or a logistic regression (LOGIT) for the presence of erosions on all patients reveal that TRAF1 markers are associated with erosions in NOAR and with the extent of the radiological damage in erosive patients in ERAS.
As expected, the results from the continuous part of the ZINB model are consistent with results from the negative binomial regression (NBREG) model, and represent the association of TRAF1 markers with the extent of Larsen score or radiological damage in erosive patients seen in ERAS (rs2900180, NBREG, Year 3: 43% increase, 95% CI 12%–81%, p = 0.004; rs10760130, NBREG, Year 3: 30% increase, 95% CI 1%–68%, p = 0.046). However, this effect is not significant in NOAR, as reported13.
The results from the susceptibility part of the ZINB model are consistent with results from the logistic regression model used to predict erosions, and represent the association of TRAF1 markers with the presence of erosions previously reported in NOAR13. However, this effect is not significant in ERAS.
The shared epitope is associated with both the extent of radiological damage and the presence of erosions in ERAS and NOAR (cross-sectional analysis: Appendix 2; longitudinal analysis: Table 2).
Combined analysis in ERAS and NOAR
To improve power, data on 445 patients from ERAS were combined with data from 1446 patients from NOAR and analyzed longitudinally. The power to detect an increase of 1 or 2 Larsen units in the combined cohort for MAF around 40% is 36% or 90%, respectively. For MAF around 10%, a difference of 2 Larsen units will be detected with a power of 80%. The results of the analysis are presented in Appendix 1. Carriage of at least 1 HLA-shared epitope allele was strongly associated with radiographic outcome in each cohort and the strength of the association was increased in the combined analysis (Table 4, ZINB combined p value: 2.81E-10, dy/dx: 4.84). A clear dose effect was observed: dy/dx for 1 copy was 4.26 and for 2 copies, 8.19. Among all SNP tested, 5 loci (TRAF1, KIF5A, PTPN22, AFF3, TAGAP) showed a ZINB combined p value < 0.05 (Table 4); however, none would remain significant after correction for multiple testing. Nevertheless, the binomial probability of obtaining, by chance, at least 5/30 associated loci at a significance level of 0.05 is 0.016. Consequently, under a prior hypo thesis of no association, there is a significant accumulation of RA-susceptibility SNP associated with radiological severity.
Results from the longitudinal zero-inflated negative binomial (ZINB) regression analysis of the Larsen score on the carriage of putative or confirmed rheumatoid arthritis susceptibility single-nucleotide polymorphisms (SNP) in the combined dataset of the Early Rheumatoid Arthritis Study (ERAS) and the Norfolk Arthritis Register (NOAR). Results are shown for SNP with a ZINB combined p value < 0.05 and for the shared epitope. All results are available in Appendix 1.
Patients carrying at least 1 copy of the minor allele of rs2900180 had on average a 2.25-unit higher Larsen score than homozygotes for the major allele (ZINB combined p value: 0.01). Adjustment for the shared epitope did not meaningfully affect the results (data not shown). No dose effect was observed: dy/dx for 1 copy was 2.59 and for 2 copies 1.40, possibly explained by a loss of power due to lower patient numbers in every category (only 410 homozygotes for the minor allele in the combined dataset).
DISCUSSION
RA susceptibility variants mapping to the TRAF1/C5 locus on chromosome 9 have previously been investigated for association with radiological outcome in patients with RA1,13,14,15. Based on recent evidence, it seems that the originally reported LD block containing rs3761847, rs10818488, and rs10760130 is not associated with radiological severity across several populations14. We reported previously an association between a neighboring LD block (rs2900180) and radiological severity13. This association had not been reported further. We have found evidence, in a second independent cohort, that this locus (TRAF1, rs2900180) is associated with Larsen score, and we have strengthened the evidence for association with this locus using cross-sectional and longitudinal modeling.
Another plausible explanation for the lack of consistency between studies is the heterogeneity in the measurement of radiological damage. For instance, in the Dutch cohort, radiological assessment is made using the Sharp-van der Heijde method, while in NOAR and ERAS, damage is measured using the Larsen score. Second, different outcome measures of radiological damage have been examined. For example, in the Dutch study, investigators focused on progression of damage over time. However, association with extent of Larsen score was investigated in both the NOAR and ERAS studies, with association reported in ERAS but not NOAR. This may reflect chance and power issues, because the sample sizes tested are modest in comparison with studies of disease susceptibility.
The longitudinal modeling techniques presented and used here offer an optimal and more powerful way to analyze time-dependent continuous traits and illustrate the nonlinear progression of radiological damage over time, the early development of erosions, the different kinetics between cohorts, and the need, if cross-sectional analyses are to be performed, to perform them at timepoints when the variance in outcome is maximal. One example would be when half of the cohort is erosive, if the outcome studied is the presence of erosions. Power calculations provided here support the fact that a longitudinal analysis is clearly a more powerful approach compared with a cross-sectional analysis, if radiographs are not performed systematically at every timepoint. The longitudinal analysis appeared to provide greater sensitivity as evidenced by the stronger statistical association observed for the HLA-SE, which served as a positive control for this method. Indeed, the p value for association between the SE and radiological damage in NOAR improves from 1.26E-07 (cross-sectional analysis, Appendix 2) to 1.46E-08 (longitudinal analysis, Table 2), and further to 2.81E-10, when the 2 cohorts are pooled.
The combined analysis, including ERAS and NOAR data, suggests several other SNP markers of severity. A replication of those results is fundamental before any conclusion can be drawn. It has been reported that the CD40 gene susceptibility risk allele confers protection for disease severity29; however, we found no evidence for association in the current datasets. Interestingly, the combined analysis identifies a total of 5 non-HLA loci of interest with a noncorrected p value for association with the Larsen score < 0.05: TRAF1, KIF5A, PTPN22, AFF3, TAGAP. This represents a significant accumulation of RA susceptibility SNP associated with radiological severity (p = 0.016).
Limitations of our study include the lack of ACPA data in the ERAS samples, which precluded an estimation of the effect of TRAF1 variants in determining radiological outcome above that accounted for by ACPA positivity. It should also be noted that no correction for multiple testing was applied in the analysis. However, if a Bonferroni corrected p value were applied to account for the number of loci tested, then association with the rs2900180 TRAF1 variants would not remain significant at the corrected threshold (p = 0.002). Further replication of these findings will therefore be required.
The lack of association between outcome and the majority of markers tested could be related to modest sample size and lack of statistical power. Alternatively, markers of RA susceptibility may not in general have the dual effect of modulating disease course. Large, multicenter collaborations are likely needed to address this issue. A well-powered, dedicated genome-wide study of disease outcome in RA remains to be reported but could identify clinically useful markers of outcome.
We report association of the rs2900180 at the TRAF1 locus with radiological damage in a second, independent cohort of patients with RA. Incorporation of longitudinal analysis techniques and combining data with that from a previous UK series increased the evidence for association with the locus still further. This confers reasonable evidence that rs2900180 is a true marker of disease severity.
Acknowledgment
The ERAS clinicians and research nurses: Dr. Paul Davies and Lynn Hill (Chelmsford), Dr. Jo Devlin, Prof. Paul Emery and Lynn Waterhouse (Birmingham), Helen Tate (Grimsby), Cathy Boys (Basingstoke), Dora White (Medway), Helen Dart (Oswestry), Sue Stafford (Winchester), Dr. John Winfield (Sheffield), Annie Seymour (St. Albans). Study coordinators/data managers: Cathy Mayes and Marie Hunt (St. Albans).
Appendix
Results from the longitudinal zero-inflated negative binomial (ZINB) regression analysis of the Larsen score on the carriage of putative or confirmed RA susceptibility single-nucleotide polymorphisms (SNP). Chr: Chromosome. Equation, severity/susceptibility: severity/susceptibility components of the ZINB model. Est: estimate, expressed as percentage increase in Larsen score for the severity part of the model, or as odds ratio (OR) for the susceptibility part of the model. Lo95: lower 95% CI. Up95: upper 95% CI. N obs: total number of observations. Z obs: among the N obs., number of observations with a Larsen score equal to zero. comb p: ZINB combined p value. dy/dx: time-averaged effect size in Larsen units (marginal effect of the model). NA: not applicable, because the SNP was either not genotyped or did not pass quality control.
Appendix
Results from the cross-sectional zero-inflated negative binomial (ZINB) regression analysis of the Larsen score on the carriage of at least 1 copy of the shared epitope single-nucleotide polymorphism (SNP).
Footnotes
-
Full Release Article. For details see Reprints/Permissions at jrheum.org
-
Supported by a core program grant from Arthritis Research UK (grant reference 17752) and by the NIHR Manchester Biomedical Research Unit. Dr. Viatte is supported by a research grant from the Swiss Foundation for Medical-Biological Scholarships (SSMBS), managed by the Swiss National Science Foundation (grant reference number PASMP3_134380). That grant is financed by a donation from Novartis to the SSMBS. ERAS grants: ARC ASG Y0514 ERAS Coordination at the Department of Rheumatology, St. Albans; 1998–2005 Project Grant Y0506, Prognostic Factors in Early RA, 1997–2001.
- Accepted for publication October 30, 2012.
Free online via JRheum Full Release option
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.