

To the Editor:
Chloroquine (CQ) has been used for the treatment and prophylaxis of malaria since World War II. Ten years later, the administration of CQ and hydroxychloroquine (HCQ) was introduced for the longterm therapy of rheumatic diseases. Although no reliable statistical data are available, HCQ, which differs from CQ only in a hydroxyl group at the end of the side chain, but not in pharmacokinetics or metabolism, is described in the literature as less toxic and has been staging a comeback, particularly in the treatment of systemic lupus erythematosus (SLE)1.
CQ and HCQ induce a dysfunction of the lysosomal enzymes, leading to the impairment of intracellular degradation processes in conjunction with the accumulation of pathological metabolic products (glycogen and phospholipids). These appear histologically as granulovacuolar cell mutations and ultrastructurally as lamellar inclusion bodies (“myeloid bodies”) and as “curvilinear bodies” in cytoplasm (remnants of poorly digested membranes)2,3. Their pharmacokinetics are characterized by a long half-life and a high volume of distribution; they follow a multicompartment model with very slow distribution between plasma and tissue, leading to sustained organ sequestration and sometimes irreversible organ damage4.
Severe toxicity in the form of irreversible retinopathy5 is well known under longterm treatment with these substances. In contrast, neuromyopathy6 and especially cardiac damage7 receive scant mention in the literature. Cardiac complications comprise conduction disturbances [bundle-branch block, atrioventricular (AV) block] and cardiomyopathy — often with hypertrophy, restrictive physiology, and congestive heart failure (literature reviews are found in Nord, et al8 and Costedoat-Chalumeau, et al9).
Because the clinical features of cardiotoxicity are unspecific, the followup of potentially affected patients is of utmost importance.
We report a case of a patient with rheumatoid arthritis (RA) and heart disease who was treated with CQ.
A 65-year-old woman was admitted for rehabilitation after having undergone surgery for lumbar spinal stenosis in April 2007. Electrocardiography revealed an atypical bundle-branch block. She reported mild dyspnea and angina pectoris (New York Heart Association Class II) for many years without progression. A diagnosis of hypertensive heart disease was rendered in 2001. In October 2006 she experienced syncope with subsequent resuscitation. The electrocardiogram showed a complete AV block, and a dual-chamber pacemaker was implanted. There were no indications of myocarditis or variation in her medications preceding the syncope. CQ medication was continued; she recovered completely.
Her history revealed that she had been diagnosed with RA 43 years before. It was treated with corticosteroids, nonsteroidal antiinflammatory drugs, and CQ. Azathioprine, methotrexate, gold compounds, and leflunomide were discontinued because of side effects. CQ had been administered for 35 years (250 mg daily), and the cumulative dose was 3195 g CQ phosphate. Except for a slow, continuous deterioration of her vision, she noted no other side effects.
Her medications upon admission included CQ phosphate 250 mg, naproxen 500 mg (twice daily), prednisolone 15 mg, oxycodone 40 mg, nebivolol 2.5 mg, torsemide 10 mg, and pantoprazole 40 mg.
On examination she appeared to be in good general condition, with blood pressure 140/80 mm Hg and heart rate 84/min. Mild synovitis of the finger joints and severe polyarthropathic deformities due to longstanding RA were noted. There was no evidence of muscular weakness, no impairment of sensation, and no diminished stretch reflexes. Heart and lungs were without pathological findings. There was no distension of the jugular veins, no edema, and vascular status was normal.
Laboratory findings showed elevation of enzyme levels (lactate dehydrogenase 426 U/l, reference −247; creatine kinase 259 U/l, ref. −145; creatine kinase-MB/mass 11.7 μg/l, ref. −3.6; troponin I 0.26 ng/ml, ref. −0.1) and rheumatoid factor (160 IU/ml, ref. −15). The remaining laboratory results were normal.
Under Holter monitoring no arrhythmia was detected; regular function of the pacemaker was noted. Transthoracic echocardiography showed normal-size atria and ventricles, borderline hypertrophy (1.2 cm), hypokinesis of the posterior and lateral wall, mitral insufficiency I–II°, diastolic dysfunction II°, and no signs of restrictive physiology.
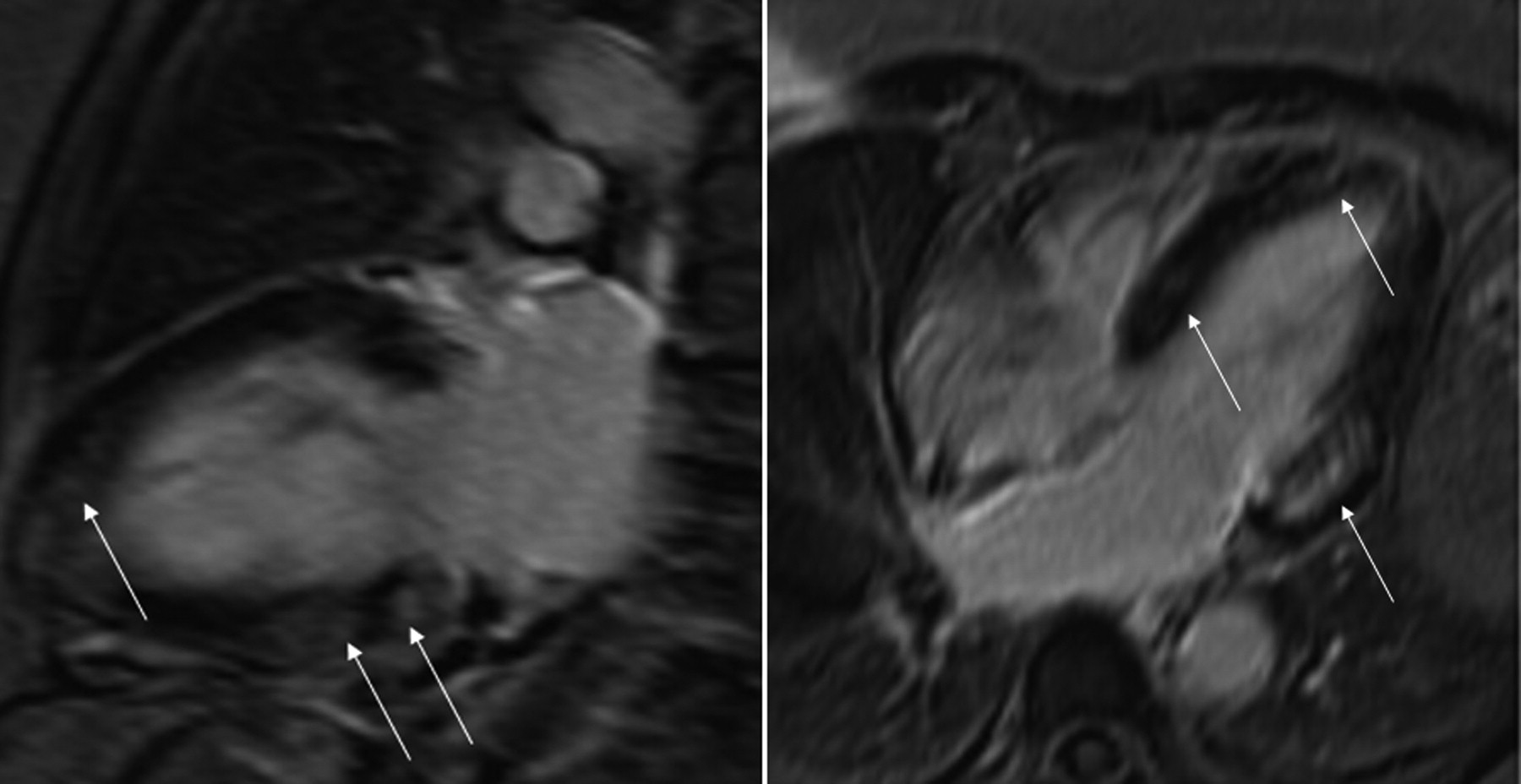
Chest radiography found the ventricular pacemaker lead in the coronary sinus, while the heart and lungs were normal. Magnetic resonance imaging (MRI) revealed left ventricular (LV) hypertrophy (lateral wall 1.9 cm) and LV ejection fraction 53%, caused by hypokinesis of the apical anterior and the basal inferolateral wall. The delayed gadolinium enhancement sequence (Figure 1) showed decreased viability in a nonvascular pattern, but no myocardial edema to suggest myocarditis.

Magnetic resonance imaging shows decreased viability of the left ventricle in a nonvascular pattern (arrows indicate delayed gadolinium enhancement). Left panel: modified 2-chamber view; right panel: 4-chamber view.
Left heart catheterization revealed a LV end-diastolic pressure of 10 mm Hg; no coronary artery disease was found. Five endomyocardial biopsies were obtained from the lateral wall, guided by the delayed enhancement pattern in MR imaging.
Pathological examination (Figure 2) showed extensive cytoplasmic vacuolization of the myocytes, mild interstitial fibrosis, no evidence of inflammation or vasculitis (including immunohistology), no amyloid deposition, and no cell necrosis. Transmission electron microscopy (Figure 3) revealed accumulation of numerous lamellar and curvilinear inclusion bodies in the myocytes. Molecular pathology (nested polymerase chain reaction) disclosed no infections of the myocardium with enteroviruses, adenoviruses, parvovirus B19, human herpesvirus 6, Epstein-Barr virus, or Borrelia burgdorferi.

Endomyocardial biopsy from the left ventricle. Light microscopy (Masson’s trichrome staining) shows marked cytoplasmic vacuolization of the myocytes with periodic acid-Schiff-positive granular deposits (insert).
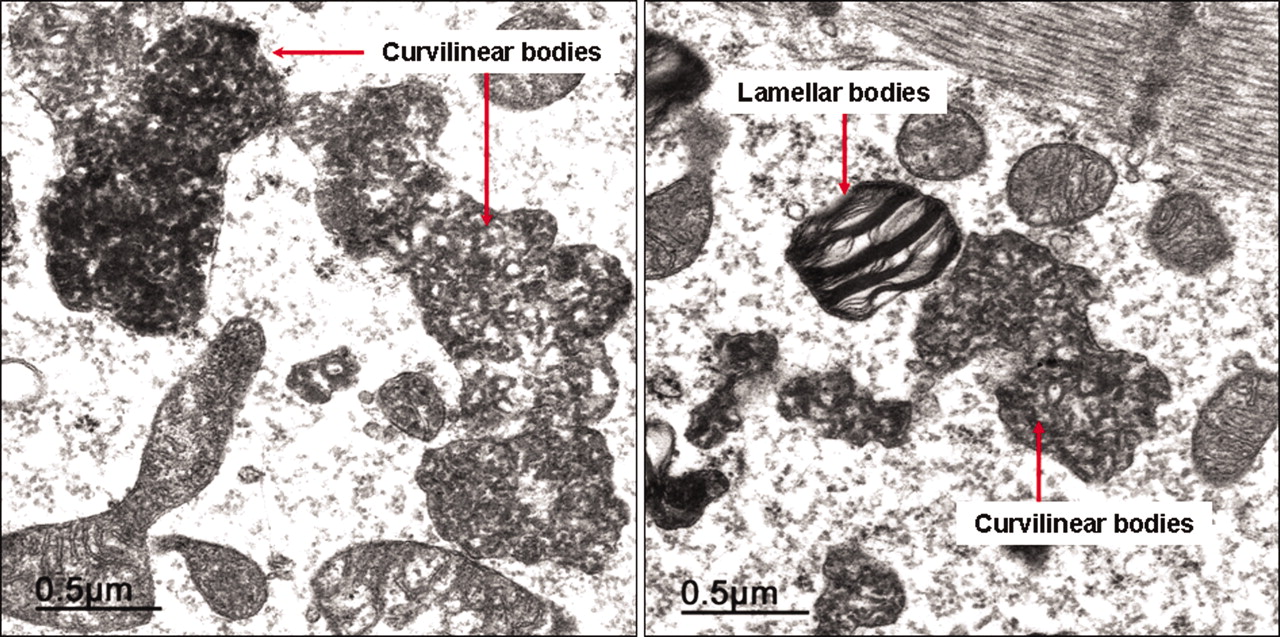
Endomyocardial biopsy from the left ventricle. Transmission electron microscopy shows lamellar and curvilinear cytoplasmic inclusion bodies in the myocytes.
The diagnosis of Fabry disease was excluded by molecular genetic testing. The serum CQ level was 0.27 mg/l, which is in the therapeutic range. Ophthalmologic examination revealed signs of chronic CQ intoxication (bull’s-eye maculopathy of the left eye).
CQ medication was discontinued. Followup 14 months later showed persistence of the complete AV block. The serum enzyme levels had decreased slightly. From a clinical point of view, there was no indication of congestive heart failure, but there was progression of retinopathy and visual loss.
As seen in our case, there are no clinical signs that indicate unambiguously that cardiotoxicity was induced by CQ. Further, there is no correlation between the different organ toxicities (cardiomyopathy, retinopathy, neuromyopathy), which would permit analogous diagnostic conclusions, and the determination of blood levels is not accurate for detecting CQ toxicity (serum level was in the therapeutic range), due to the complex pharmacokinetics and the large interindividual fluctuation range in the metabolism of the substance4. Thus, the cardiac process may be asymptomatic for a long period.
Because of the small number of cases and the lack of systematic studies, it is impossible to calculate the incidence of CQ cardiotoxicity; however, a considerable number of undetected cases can be assumed. To date, reports have been published on 35 patients with biopsy-proven cardiomyopathy7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36 and on 30 patients with total AV block only9,30,37,38. Moreover, 12 of those patients with cardiomyopathy exhibited complete AV block as well (Table 1). Most patients were treated with CQ, but in recent years there has been a clear trend toward HCQ. All except one of them (who had recurrent malaria) had connective tissue diseases (predominantly RA and SLE).
Details of chloroquine/hydroxychloroquine-induced cardiomyopathies (case reports including histologic studies of the myocardium).
The cumulative dose of CQ/HCQ (15–5040 g) and the duration of treatment (2–31 years) vary greatly. Our patient had the longest duration of treatment (35 years) and one of the highest cumulative doses (3195 g).
To date, it has not been possible to establish predictive values for any of the measures, such as age, sex, duration of treatment, cumulative dose, or underlying illness, in order to estimate toxicity in individual cases.
Cardiac complications include conduction disturbances and cardiomyopathy. As in our patient, several cases have been described in the literature in which complete AV block precedes the cardiomyopathy and the medication continues to be administered because of improper evaluation of the pathogenesis9,15,23. We could not determine other causes for the AV block, and there is no known accumulation of higher-grade AV conduction disorders in conjunction with RA39. Recently, the case of a patient with sick sinus syndrome as a result of CQ therapy was also reported32.
The cardiomyopathy is predominantly of the restrictive type (with diastolic dysfunction), but mainly systolic impairment has also been reported (Table 1). The most common pathological result in cardiac imaging is myocardial hypertrophy. Congestive heart failure is the prevailing clinical symptom. However, some patients have very few, discrete symptoms, or none at all (as in our case, in which we found no indication of restrictive kinetics, either).
In view of the large number of variables in the clinical hypothesis, the histological examination plays an extremely important role. As a rule, the morphology of the tissue damage caused by CQ/HCQ is identical in all organs involved. Light microscopic study reveals a vacuolar myopathy. Ultra-structurally, lamellar and so-called “curvilinear” inclusion bodies can be demonstrated. The curvilinear bodies are comma-shaped structures which in human pathology occur only in cases of CQ damage and in neuronal ceroid lipofuscinosis, a group of hereditary neurodegenerative disorders in children7,8,40.
While curvilinear bodies definitively document the pathogenesis of CQ toxicity, the presence of lamellar inclusion bodies is also described after the administration of amiodarone and in other storage diseases41 (Table 2). In particular, Fabry disease, which is also accompanied by myocardial hypertrophy, can make the differential diagnosis difficult20,28.
Differential diagnoses of chloroquine/hydroxychloroquine cardiomyopathy.
The range of differential diagnoses in the case of vacuolar myopathy revealed under light microscopy is considerably larger and includes the connective tissue diseases and steroid myopathy2,3 (Table 2). Because rheumatic diseases often involve the cardiovascular system, and cortisone preparations are frequently prescribed, overlaps can occur and the symptoms of toxic damage are often ascribed to the underlying rheumatic disease and its complications.
To avoid the sampling error of blind biopsies (because of a patchy distribution of the pathological process in the myocardium) we took MR-guided biopsies from the left ventricle, following the delayed enhancement pattern in MRI42. Both the vacuolar myopathy (Figure 2) as well as the curvilinear bodies (Figure 3) were unequivocally documented in the myocardial specimen, so that the diagnosis of CQ cardiotoxicity was definitive. Further, the following diseases were excluded by means of differential diagnosis and testing: coronary artery disease, myocarditis and vasculitis (because of the underlying rheumatic disease), viral myocarditis, amyloidosis, steroid myopathy, and Fabry disease.
Based on strict histological (ultrastructural) criteria, the diagnosis of CQ/HCQ-induced cardiomyopathy has been confirmed pathologically in only 24 patients to date (Table 1).
The prognosis for patients with this cardiomyopathy appears to be mixed: heart transplants had to be performed in 2 patients with refractory heart failure9,22. Thirteen patients died (most of them soon after the diagnosis was established). The course of the illness was not clear for 8 patients, and an improvement was described in only 10 cases (Table 1).
It must be noted that cardiotoxicity is difficult to diagnose under longterm treatment with CQ/HCQ and is presumably often overlooked in everyday clinical practice. Early detection is of utmost importance, as no therapy is available, the reversibility of organ damage is questionable, and severe courses of illness have been described. In case of longterm treatment and high cumulative doses, we propose at least annual followup examinations (including electrocardiography). If myocardial damage is suspected, further diagnostic measures including ultrastructural examination of myocardial tissue are required.
Acknowledgment
The authors thank Dr. M. Farr (Department of Cardiology, Heart and Diabetes Center NRW, Ruhr University of Bochum, Bad Oeynhausen, Germany) for conducting the molecular genetic testing for Fabry disease.








{kind=link}
{kind=link}
{kind=link}