

Abstract
Objective. To evaluate efficacy and safety of CE-224,535, a selective P2X7 receptor antagonist, versus placebo, in patients with active rheumatoid arthritis (RA) and inadequate response to methotrexate (MTX).
Methods. In our phase IIA study (ClinicalTrials.gov no. NCT00628095; A6341009), patients aged ≥ 18 years with active RA were randomized to receive either CE-224,535 (500 mg bid) or placebo for 12 weeks; all patients continued a stable background dose of ≥ 7.5 mg MTX.
Results. The American College of Rheumatology 20% (ACR20) response rate (primary efficacy endpoint) was not significantly different from placebo for CE-224,535 (34.0% vs 36.2%; p = 0.591) at Week 12, or at any timepoint over the 12-week treatment period. There was no significant difference at Week 12 for the ACR20 response rate following subgroup analyses by age, sex, baseline disease activity, baseline duration of disease, geographic region, or concomitant use of steroids. ACR50/ACR70 response rates and change from baseline in Disease Activity Score 28-joint C-reactive protein (DAS28-3-CRP) and Health Assessment Questionnaire-Disability Index for CE-224,535 were not significant at Week 12 versus placebo. Treatment-emergent adverse events (AE) were reported by 62.3% (CE-224,535) and 55.3% (placebo) of patients; the most common AE were nausea (11.3%, CE-224,535; 4.3%, placebo) and diarrhea (7.5%, CE-224,535; 4.3%, placebo). The proportion of patients discontinuing due to an AE was 9.4% (CE-224,535) and 6.4% (placebo); no deaths were reported. Serious AE occurred in 3.8% (CE-224,535) and 2.1% (placebo) of patients; none was considered treatment-related.
Conclusion. CE-224,535 was not efficacious, compared with placebo, for the treatment of RA in patients with an inadequate response to MTX. CE-224,535 demonstrated an acceptable safety and tolerability profile.
Rheumatoid arthritis (RA) is a chronic, progressive autoimmune disease characterized by joint inflammation and destruction, resulting in decreased quality of life1. Current treatment options for RA include traditional disease-modifying antirheumatic drugs (DMARD), such as methotrexate (MTX), and biologic response modifiers2,3,4,5.
The ultimate goal of treatment for this incurable condition is to attain disease remission (no disease activity)6. However, an inadequate number of patients achieve an adequate treatment outcome, partly due to the lack of a consensus definition of remission6 and the absence of clear guidelines on the use and interpretation of disease activity assessment tools, leading to inconsistent clinical practice7. Also, some patients do not respond to available treatments or they develop intolerable side effects to them, therefore additional treatment options using a unique mechanism of action are desirable8.
The purinergic P2X7 receptor is an adenosine triphosphate (ATP)-gated ion channel found on cells of the hematopoietic lineage, including lymphocytes, monocytes, and macrophages9. It is an important cell-surface regulator of several key inflammatory molecules10, and ATP activation initiates increased posttranslational modification of interleukin 1 (IL-1) and IL-18, activating both their functionality and secretion11,12,13,14,15. IL-1 has been strongly implicated in the multiple inflammatory pathways involved in the pathogenesis of RA16. A preclinical study demonstrated that key catabolic events are mediated by IL-18 signaling in human articular chondrocytes17. In addition, a recent study showed that both the expression levels and biological activity of IL-18 in the serum, synovial fluid, and tissue of patients with RA were significantly increased compared with samples from healthy adults18, suggesting a role for IL-18 signaling pathways in RA.
The biologic therapy anakinra, an IL-1 receptor antagonist, has demonstrated modest efficacy in patients with RA19,20,21. However, the pharmacokinetic/safety profile associated with this treatment is not optimal20,22,23,24; more frequent dosing may increase patient benefits, yet remains impractical due to the properties of the drug and administration method (daily injection).
CE-224,535 was developed as a DMARD, and is a selective antagonist of the human P2X7 receptor, reducing leukocyte secretion of IL-1ß and IL-18, thereby providing a novel therapeutic approach for treatment of RA. It is possible that more robust efficacy could be achieved with upstream inhibition of IL-1 rather than the current IL-1 receptor inhibition; it was therefore anticipated that CE-224,535 had this potential, given its inhibition of posttranslational processing and release of IL-1ß and IL-1825,26.
Our phase IIA study investigated the efficacy and safety of CE-224,535 in patients with active RA inadequately controlled by MTX.
MATERIALS AND METHODS
Our study (A6341009) was sponsored by Pfizer Inc. and was conducted in 24 centers in 7 countries (Chile, Czech Republic, Mexico, Poland, Republic of Korea, Spain, and the United States). The trial is registered with ClinicalTrials.gov, number NCT00628095. The final protocol, any amendments, and informed consent documentation were reviewed and approved by the institutional review boards or the independent ethics committees at each of the participating investigational centers. In addition, our study was conducted in compliance with the ethical principles originating in or derived from the Declaration of Helsinki and in compliance with all International Conference on Harmonization Good Clinical Practice Guidelines.
Patients
Eligible patients were aged ≥ 18 years with a diagnosis of active RA based on the American College of Rheumatology (ACR) 1987 revised criteria27, despite ongoing MTX treatment. Active disease was defined as ≥ 4 tender/painful joints on motion and ≥ 4 swollen joints (28-joint count). Patients also had to meet ACR 1991 Revised Criteria for Global Functional Status in RA, Class I, II, or III.
Ongoing MTX treatment was required at a weekly dose of ≥ 7.5 mg (oral or parenteral) for 3 months prior to study entry, at a stable dose for ≥ 4 weeks prior to screening, and taken with concomitant therapy (folic acid at a minimum of 400 μg daily, or equivalent dosing). Other DMARD permitted included sulfasalazine, leflunomide, injectable or oral (auranofin) gold, D-penicillamine, chloroquine, or hydroxychloroquine. No more than 2 of these DMARD, in addition to MTX, were to be given during the study or within 1 month prior to randomization.
Patients were excluded from the study if they had any other inflammatory arthritis that could have interfered with disease activity assessments, tuberculosis without treatment and/or positive tuberculin reaction without known vaccination, 12-lead electrocardiogram demonstrating QTc > 460 ms for men and > 480 ms for women at screening, or any condition that could possibly have affected oral drug absorption.
In addition, patients were excluded if they had participated in previous CE-224,535 studies, or received the following treatments: pharmacologically active herbal supplements; corticosteroids by any route with the exception of a stabilized oral dose of ≤ 10 mg prednisone or equivalent/day (which was allowed); and the following medications given at the specified intervals prior to study treatment: azathioprine, cyclosporine, anakinra, or etanercept (≤ 4 weeks); infliximab, adalimumab, or any experimental RA therapy (≤ 8 weeks); abatacept (≤ 3 months); or rituximab (≤ 12 months). Patients were also excluded if they had a history of chronic or serious life-threatening infection, severe, progressive and/or uncontrolled renal, hepatic, hematological, gastrointestinal, endocrinological, pulmonary, cardiac, or neurological disease.
Study design
Our phase IIA, randomized, double-blind, placebo-controlled, parallel-group, multicenter study investigated CE-224,535 in the treatment of the signs and symptoms of RA in patients inadequately controlled by MTX (Figure 1). All patients were to continue a stable background weekly dose of at least 7.5 mg MTX during the study.

Study design. Patients continued a stable background weekly dose of at least 7.5 mg methotrexate during the study. bid: twice daily; BL: baseline (randomization).
The primary objective was to test the efficacy of CE-224,535 compared with placebo as assessed by the ACR20 response rate at Week 12. The secondary objectives of the study were to evaluate the safety and tolerability of CE-224,535, the pharmacokinetic profile of CE-224,535, and health and functional status.
Treatment
Patients were randomized equally to receive either oral CE-224,535, 500 mg twice daily (bid), or placebo for 12 weeks.
Rescue medication
Acetaminophen was allowed as rescue medication if dosed ≤ 2.6 g/day for ≤ 4 consecutive days. If a patient was already taking stable background doses of acetaminophen, they could increase the dose up to 2.6 g/day for up to 4 consecutive days for rescue purposes. For patients who were not on stable, background opioid therapy, any of the following single opioid agents could be given as rescue medication (with or without acetaminophen) for ≤ 4 consecutive days up to a maximum potency equivalent to 30 mg/day of orally administered morphine: hydrocodone, not exceeding 30 mg total daily dose; oxycodone, not exceeding 15 mg total daily dose; or tramadol, not exceeding 300 mg total daily dose.
Study assessments
The primary efficacy endpoint was ACR20 response rate at Week 12. ACR20 response rate was also assessed at Weeks 2, 4, and 8. In addition, the differential effects of region, sex, age, duration of disease, disease activity at baseline, and concomitant steroid use were examined for ACR20 response rate at Week 12. ACR50, ACR70, Disease Activity Score (DAS)28-3[C-reactive protein (CRP)], Health Assessment Questionnaire Disability Index (HAQ-DI), and components of the ACR response were all assessed at randomization and Weeks 2, 4, 8, and 12 (or early termination).
Blood samples for pharmacokinetic analysis were collected prior to dosing at the randomization visit and at least 2 hours after dosing; at Week 2, 2 samples were taken after dosing, with the second sample collected at least 2 hours after the first sample but prior to the next 12-hour dosing; at Week 4, 2 samples were collected: the first prior to dosing and the second at least 3 hours after dosing. Plasma samples were analyzed for CE-224,535 concentrations using a validated, sensitive, and specific high-performance liquid chromatography tandem mass spectrometric method; concentrations were summarized for each of the sampling timepoints.
Safety was evaluated throughout the study by monitoring adverse events (AE), laboratory evaluations, physical examinations, vital signs, and electrocardiograms. All patients who received study treatment were analyzed for AE; 1 patient in the CE-224,535 group was not analyzed for laboratory data because that patient discontinued the study before postdose laboratory assessments were performed.
Statistical analyses
The null hypothesis was that there was no difference between CE-224,535, 500 mg bid and placebo on the percentage of ACR20 responders at Week 12. The alternative hypothesis was that the patients treated with CE-224,535, 500 mg bid had at least a 25% higher response rate in ACR20 than the placebo group at Week 12. Categorical variables (ACR20) were analyzed by chi-square test, unless the normal approximation to the binomial distribution was not appropriate. If this was the case, Barnard’s exact test was to be used.
Baseline values were included as covariates in the analysis of covariance model used in the analysis of secondary efficacy endpoints. The differential effects of sex, age, region, duration of disease, disease activity, and their interaction with treatment were examined in the Week 12 analyses of the primary efficacy variable (ACR20). Duration of disease was dichotomized as ≤ 5 years versus > 5 years. Age was dichotomized as ≤ 65 years versus > 65 years. No pharmacokinetic analysis was conducted; pharmacokinetic data were summarized descriptively. All the safety data were summarized descriptively by randomized treatment group through appropriate data tabulations and descriptive statistics. The safety endpoints were evaluated by comparing CE-224,535, 500 mg bid to placebo using sponsor data standards.
The full analysis set (FAS) included all patients who received ≥ 1 dose of the randomized study treatment; the safety analysis set equaled the FAS. For the primary endpoint, 3 data analyses were performed for robustness: last observation carried forward (LOCF), nonresponder imputation (NRI), and observed cases (no imputation). Throughout this report, data are presented for LOCF analyses only.
RESULTS
Patient disposition
Overall, 138 patients were screened, of which 100 were assigned to study treatment: 53 received CE-224,535, 500 mg bid; 47 received placebo. The proportion of patients completing the study was 86.8% (CE-224,535, 500 mg bid) and 85.1% (placebo; Figure 2). Seven patients (13.2%) in the CE-224,535 group and 7 patients (14.9%) in the placebo group discontinued from the study. Three patients (5.7%) in the CE-224,535 group and 3 patients (6.4%) in the placebo group discontinued due to AE related to the study treatment, and 3 patients (6.4%) in the placebo group discontinued due to lack of efficacy (Figure 2). Four patients (7.5%) in the CE-224,535 group discontinued for reasons not related to the study treatment: 2 (3.8%) discontinued due to non-treatment-related AE and 2 (3.8%) discontinued because they were no longer willing to participate. One patient (2.1%) in the placebo group discontinued for reasons not related to the study treatment (Figure 2).

Patient disposition. AE: adverse event; bid: twice daily; LOE: lack of efficacy; NLW: no longer willing to participate in study.
Baseline demographics and patient characteristics
Demographic characteristics were comparable between the treatment groups. Patients were aged between 21 and 78 years and the majority were white and female (Table 1). The mean duration since first diagnosis of RA was 7.5 years (placebo) and 8.7 years (CE-224,535). The mean duration of treatment was similar between treatment groups: 85 days (placebo) and 84 days (CE-224,535). The mean MTX dose at baseline was 13.8 mg and 15.5 mg in the CE-224,535, 500 mg and placebo groups, respectively. The proportions of patients on concomitant glucocorticoids were 67.9% and 66.0% in the CE-224,535, 500 mg and placebo groups, respectively. The proportions of patients receiving concomitant DMARD were 20.8% and 19.1%, respectively.
Baseline demographics/patient characteristics.
Efficacy
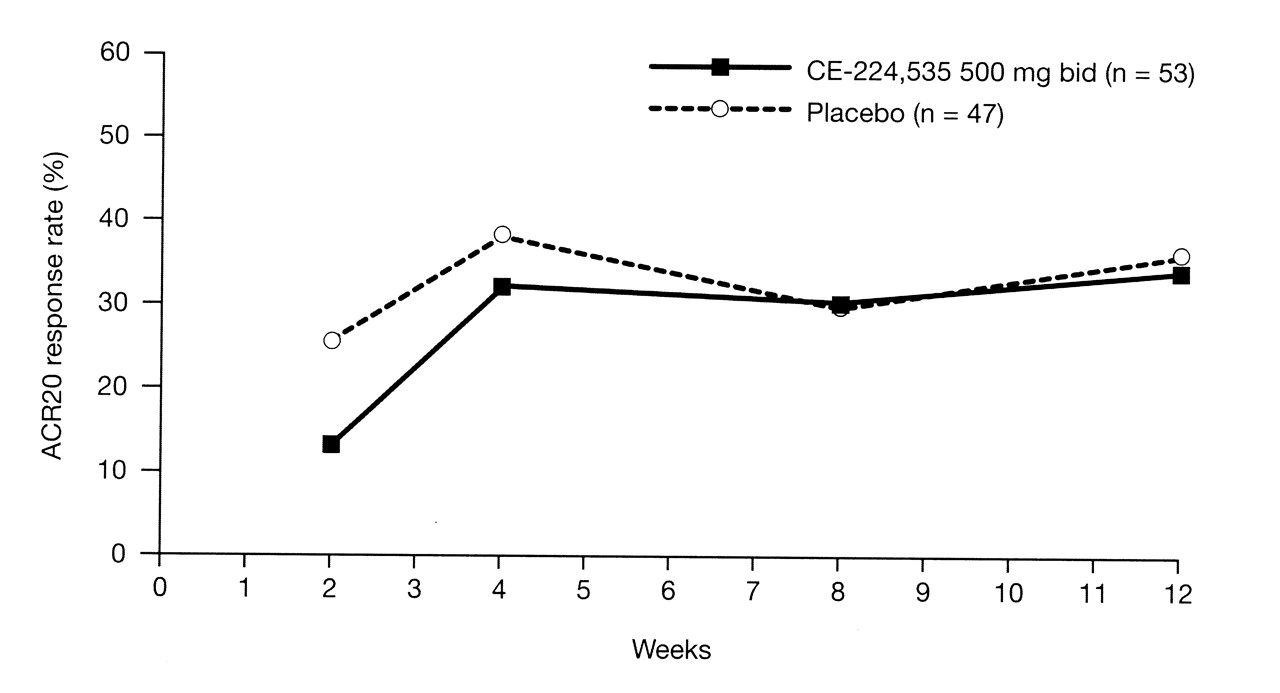
There was no significant difference in ACR20 response rate at Week 12 (primary efficacy endpoint) between CE-224,535 (34.0%) and placebo [(36.2%; 90% CI −0.17, 0.13; p = 0.591 (FAS, LOCF)]. The ACR20 response rate was not significant at any timepoint over the 12-week study period (Figure 3). The ACR20 analysis using NRI was consistent with that of LOCF; ACR20 response rates at Week 12 using NRI were 30.2% and 36.2% for CE-224,535 and placebo, respectively (90% CI −0.21, 0.09). There were no significant changes from baseline for CE-224,535 versus placebo in any of the ACR component scores throughout the study period.

ACR20 responder rate over 12 weeks; primary endpoint was ACR20 response rate at Week 12 (full analysis set; last observation carried forward). There was no significant difference between CE-224,535 and placebo at any timepoint. ACR: American College of Rheumatology; bid: twice daily.
No statistically significant difference was observed between CE-224,535 and placebo in the ACR20 response rate following subgroup analyses at Week 12 by age, sex, baseline disease activity, baseline duration of disease, region, or concomitant use of steroids (LOCF, FAS; Table 2). Week 12 ACR20 response rates for CE-224,535 trended higher in those patients with higher baseline CRP levels compared to those for placebo. ACR20 response rates were 50.0% and 47.4% for baseline CRP ≥ 6 mg/l and ≥ 8 mg/l, respectively, in the CE-224,535 group compared with 26.1% and 30.0% in the placebo group (Table 2). ACR50 and ACR70 responses were not significant at Week 12: 11.3% and 17.0% for CE-224,535 and placebo groups, respectively (p = 0.793; ACR50); 3.8% and 0% for CE-224,535 and placebo groups, respectively (p = 0.121; ACR70; Table 2).
Secondary efficacy endpoints at Week 12: ACR20 response rates by age, sex, baseline disease activity, baseline duration of disease, region, or concomitant use of steroids; ACR50; ACR70; DAS28-3-CRP; HAQ-DI (full analysis set; last observation carried forward).
At Week 12, the least-squares mean changes from baseline in DAS28-3(CRP) were −0.9 and −0.8 for CE-224,535 and placebo groups, respectively (p = 0.752), and in HAQ-DI were −0.2 and −0.3 for CE-224,535 and placebo groups, respectively (p = 0.784; Table 2).
No significant difference was observed for either physician’s global assessment of arthritis or patient’s global assessment of arthritis.
Safety. Adverse events
The number of all-causality AE reported was similar between treatment groups: 72 (CE-224,535) and 61 (placebo); the number of treatment-related AE was 34 (CE-224,535) and 29 (placebo). The percentage of patients reporting all-causality AE was similar between treatment groups: 62.3% (CE-224,535) and 55.3% (placebo); the percentage of patients reporting treatment-related AE was 34.0% (CE-224,535) and 31.9% (placebo). Incidence of AE in 3 or more patients per treatment group is presented in Table 3. The most frequently reported all-causality AE in the CE-224,535 treatment group were nausea (11.3%) and diarrhea (7.5%); in the placebo group, the most common AE were headache (8.5%), upper abdominal pain, insomnia, increased alanine aminotransferase (ALT), and increased aspartate aminotransferase (all 6.4%).
Most frequently reported adverse events (safety analysis set).
The percentage of patients reporting all-causality serious AE (SAE) was 3.8% and 2.1% in the CE-224,535 and placebo groups, respectively; none was considered treatment-related. The percentage of patients reporting severe AE was 9.4% (CE-224,535) and 4.3% (placebo); only 1 patient’s AE of abdominal pain/tenderness was considered treatment-related (in the CE-224,535 treatment group). In the CE-224,535 group, 1 patient had SAE of pelvic fracture, capsular contracture associated with breast implant, and breast prosthesis implantation, and 1 patient had an SAE of depression. In the placebo group, 1 patient had SAE of road traffic accident, back injury, and contusion. None of these SAE was considered treatment-related.
Discontinuations (all-causality) due to AE occurred in 9.4% and 6.4% of patients in the CE-224,535 and placebo groups, respectively (Figure 2). Reasons for permanent discontinuation included vomiting, abdominal pain, and arthralgia (CE-224,535), and moderate worsening of RA, arthralgia, and increased ALT (placebo). There were no deaths reported throughout our study.
Infections
The incidence of infections was similar between treatment groups: 24.5% (CE-224,535) and 21.3% (placebo); the most common were bronchitis (5.7%, CE-224,535; 2.1%, placebo) and nasopharyngitis (5.7%, CE-224,535; 2.1%, placebo).
Abnormal laboratory findings
There were no notable changes from baseline or between the treatment groups in median changes from baseline for any laboratory measurement. There were no notable differences between treatment groups in the numbers of patients with laboratory abnormalities after dosing. All clinical laboratory-related treatment-emergent AE were of mild or moderate severity and none was reported as an SAE; leukopenia, lymphopenia, and neutropenia occurred in 1.9% of patients in the CE-224,535 treatment group and thrombocytopenia occurred in 2.1% of patients in the placebo group. Transaminase elevations were recorded only in the placebo group (Table 3).
Pharmacokinetics
The median trough concentration of CE-224,535 in our study was ∼250 ng/ml; this concentration is about 25 times the estimated 90% inhibitory concentration (IC90) for inhibition of IL-1ß release as determined previously in an ex vivo whole-blood assay. Patients whose baseline CRP was ≥ 8 mg/l (n = 17) had median trough concentrations about twice as much as those with baseline CRP < 8 mg/l (n = 27). A relationship between exposure and clinical response could not be inferred despite the concentrations adequately exceeding the IC90 estimate.
DISCUSSION
Our objective was to test the efficacy of CE-224,535 compared with placebo, as assessed by the ACR20 response rate at Week 12 in patients with RA who were inadequately controlled taking MTX. Our study demonstrated that CE-224,535 was not effective compared with placebo for the primary efficacy endpoint, ACR20 response rate at Week 12, or any of the secondary efficacy variables, despite adequate drug exposure. When investigating treatments for currently incurable, chronic, and complex diseases such as RA, it is essential to consider all potential therapeutic targets, particularly those with a novel mechanism of action. Although the P2X7 receptor appears to operate as an important element of the proinflammatory cascade, and antagonists of its function are expected to induce antiinflammatory outcomes, our study suggests that inhibition of the P2X7 receptor was unsuccessful in improving the signs and symptoms of RA.
One potential explanation for the lack of treatment effect is the complex nature of the pathogenesis of RA. Activation of the P2X7 receptor by ATP triggers the maturation and subsequent release of IL-1 and IL-1811,12,13,14. However, it would seem that inhibition of the release of IL-1 and IL-18 alone is insufficient to substantively mitigate the pathogenesis and maintenance of inflammation in RA. This is likely due to the presence of multiple deregulated pathways that produce additional key proinflammatory cytokines28.
It is also worth considering the patient population and whether this was a contributing factor to the study findings. Although the disease activity entry criteria (i.e., joint counts and acute-phase reactant thresholds) were not generally as rigorous as those often used in clinical studies investigating similar endpoints in patients with RA29,30, the baseline disease activity of the patients enrolled in the study was sufficient to enable the detection of an antiinflammatory effect.
Although all patients remained on a stable background weekly dose of MTX during the study, it was demonstrated in a previous study that there is no drug-drug interaction (DDI) between CE-224,535 and MTX (data on file, Pfizer Inc.). It is therefore unlikely that concomitant administration of MTX and CE-224,535 affected the potential efficacy of either drug. The observed plasma concentrations of CE-224,535 in our study were similar to those previously reported in the DDI study with MTX, and the exposures (over 24 hours) adequately exceeded the estimated IC90 (10 ng/ml) for inhibition of IL-1ß release in a multiple-dose tolerance study (data on file, Pfizer Inc.). It is possible that the lack of efficacy observed in this study was due to insufficient receptor occupancy in vivo. However, this is unlikely, as the CE-224,535 plasma concentrations achieved in the study were much greater than the concentration required to inhibit IL-1 secretion ex vivo (25 times the estimated IC90 for inhibition). Assuming that plasma concentrations were sufficient to achieve inhibition of the P2X7 receptor, it appears that blockade of the P2X7 receptor alone is not as effective as IL-1 receptor inhibition at mediating the inflammatory effects of IL-1ß. Trough concentrations were higher in patients with CRP levels ≥ 8 mg/l compared with those with CRP levels < 8 mg/l. During inflammation, hepatic drug-metabolizing CYP enzymes, particularly CYP3A4, are downregulated (likely due to IL-6 action), thus decreasing clearance and increasing the levels of drugs administered31,32. Since CE-224,535 is thought to be extensively metabolized by CYP3A (< 20% of the dose excreted unchanged in the urine), it is expected that the underlying inflammation (as reflected in CRP measurements) will cause an alteration of hepatic metabolism and lead to differences in drug concentrations.
It is also unlikely that treatment compliance was an issue in our study; the median duration of treatment with the study drug was 84 days (similar to placebo) and the protocol stipulated that treatment compliance had to be a minimum of 70%. One point for consideration is that if discontinuations occurred early in the study period, these patients would have discontinued before they had a chance to improve; therefore use of LOCF imputation would result in scores closer to baseline values being carried forward.
A dose of 500 mg bid was chosen for the study as it was well tolerated and provided the potential to test the pharmacokinetic coverage-based efficacy hypothesis. This dose was expected to result in concentrations exceeding the IC90 by more than 10-fold for the entire dosing period. The 500 mg bid dose appeared to be safe and well tolerated in patients with RA who were inadequately controlled by MTX. There was no notable difference between treatment groups in the proportion of patients with all-causality and treatment-related AE or SAE. Further, the proportion of discontinuations was comparable between treatment groups.
CE-224,535 appeared to be generally safe and well tolerated, but did not demonstrate a statistically significant separation from placebo in any efficacy outcome variable. Thus, it is probable that inhibition of the P2X7 receptor is not an efficacious strategy for the management of RA.
Acknowledgment
The authors thank the A6341009 investigators and study team, and also Jennie Frain, PhD, from Complete Medical Communications, who provided medical writing assistance funded by Pfizer Inc.
Footnotes
-
Supported by Pfizer Inc. C.A. Mebus, X. Wang, P. Gupta, and T.C. Stock own stock in Pfizer Inc. Dr. Wei has received research grants from Pfizer Inc.
- Accepted for publication December 1, 2011.








{kind=link}
{kind=link}
{kind=link}