
Abstract
Objective. Glucosamine sulfate (GS) has been inferred to have a potential antiinflammatory effect on osteoarthritis (OA). We investigated its effect on prostaglandin E2 (PGE2) in human OA chondrocytes, and the level in the PGE2 pathway at which its effect takes place.
Methods. We investigated the effect of GS treatment (0.05, 0.2, 1.0, and 2.0 mM) in OA chondrocytes in the absence or presence of interleukin 1ß (IL-1ß; 100 pg/ml). We determined the expression levels and protein production/activity of PGE2, cyclooxygenase-1 (COX-1), COX-2, microsomal PGE synthase-1 (mPGES-1), glutathione, and peroxisome proliferator-activated receptor-γ (PPARγ), using specific primers, antibodies, and assays.
Results. GS treatment at 1 and 2 mM significantly inhibited (p ≤ 0.03) production of endogenous and IL-1ß-induced PGE2. GS in both the absence and presence of IL-1ß did not significantly modulate COX-1 protein production, but GS at 1 and 2 mM demonstrated a decrease in COX-2 glycosylation in that it reduced the molecular mass of COX-2 synthesis. Under IL-1ß stimulation, GS significantly inhibited mPGES-1 messenger RNA expression and synthesis at 1 and 2 mM (p ≤ 0.02) as well as the activity of glutathione (p ≤ 0.05) at 2 mM. Finally, in both the absence and presence of IL-1ß, PPARγ was significantly induced by GS at 1 and 2 mM (p ≤ 0.03).
Conclusion. Our data document the potential mode of action of GS in reducing the catabolism of OA cartilage. GS inhibits PGE2 synthesis through reduction in the activity of COX-2 and the production and activity of mPGES-1. These findings may, in part, explain the mechanisms by which this drug exerts its positive effect on OA pathophysiology.
- GLUCOSAMINE SULFATE
- OSTEOARTHRITIS
- PROSTAGLANDIN E2
- MICROSOMAL PGE SYNTHASE-1
- CYCLOOXYGENASE
Osteoarthritis (OA) is the most common form of arthritis and is characterized by degradation and loss of articular cartilage. The hallmark of the disease is the progressive degeneration of articular cartilage and subsequent joint space narrowing. OA leads to pain, loss of motion, instability, and physical disability, thus impairing quality of life. OA results from a complex system of interacting mechanical, biological, biochemical, molecular, and enzymatic/inflammatory feedback loops. The final common pathway is joint tissue destruction resulting from failure of cells to maintain a homeostatic balance between matrix synthesis and degradation. As the disease advances, the degradative process eventually exceeds the anabolic process, leading to progressive joint tissue lesions.
Prostaglandin E2 (PGE2) is implicated in joint inflammation and is among the major catabolic mediators involved in cartilage degradation1,2,3. OA cartilage spontaneously releases more PGE2 than normal cartilage4 and knockout mice for a PGE2-specific receptor demonstrated absence of cartilage degradation in a collagen-induced arthritis model5. Prostaglandins are biologically active metabolites of arachidonic acid. PGE2 is a well-characterized mediator of inflammation. It contributes to the pathogenesis of arthritis not only by inducing pain, but also by increasing the production of catabolic molecules including proinflammatory cytokines, matrix metalloproteinases (MMP), and reactive oxygen species, which in turn contribute to articular tissue alterations. It is believed that several of the effects of interleukin-1ß (IL-1ß) are associated with stimulation of production of PGE2. In addition to exerting inflammatory actions of its own, PGE2 can potentiate the effects of other mediators of inflammation.
An antiinflammatory potential has been inferred from the effect of glucosamine sulfate (GS) through PGE2, which can explain, at least in part, the significant effect of GS in several short- and longterm clinical trials in OA, some even showing structure-modifying effects in patients with knee OA7,8,9,10,11. This contrasts with another recent report that glucosamine hydrochloride (G.HCl) did not reduce pain effectively in patients with OA12. Although G.HCl is not considered effective in contrast to GS13, when combined with chondroitin sulfate in the latter study12, it provided statistically significant pain relief compared with placebo in a subset of patients with moderate to severe knee pain. A possible explanation for this could be that patients with more severe OA experience increased inflammation, which permits different glucosamine preparations to exhibit their effectiveness.
The effect of GS on chondrocytes and cartilage has been investigated primarily in animal studies, with very few studies investigating its effect on human OA cells. In brief, GS was shown to prevent cartilage degradation in animals by acting directly on chondrocytes. It prevented proteoglycan degradation14,15, an effect that is likely secondary to the upregulation of aggrecan gene expression and downregulation of aggrecanase16,17,18. It also downregulates members of the metalloproteinase family16,17,19 and exerts antiinflammatory activities by diminishing nitric oxide and PGE2 levels20,21. The latter effects could be due to inhibition of nuclear factor-κB (NF-κB) activation by GS22, which is suggested to be a major mechanism of action of GS in OA23.
To date, the clinical efficacy of drugs targeting inhibition of PGE2 has been limited to the inhibition of activities of cyclooxygenase (COX)-1 and COX-2 (nonsteroidal antiinflammatory drugs; NSAID) or COX-2 selective inhibitors (COXIB). Although COXIB were associated with reduced incidence of the serious gastrointestinal side effects observed with conventional NSAID, these drugs demonstrated increased risk of cardiovascular events associated with inhibition of COX-2. For this reason, rofecoxib was withdrawn worldwide in late 2004, followed by valdecoxib in 2005. This withdrawal has raised several serious questions regarding the balance of safety and efficacy of this class of drug and as a result, alternative methods for PGE2 inhibition with no serious side effects are crucially needed; GS is potentially one such compound.
We investigated, in OA chondrocytes, the effect of GS treatment on the level of PGE2 as well as on precursor enzymes responsible for its production.
MATERIALS AND METHODS
Specimen selection
OA specimens were obtained from the femoral condyles and tibial plateaus of patients undergoing total knee arthroplasty (13 women and 8 men; mean age 71 ± SD 10 yrs). All patients were evaluated as having OA according to the American College of Rheumatology clinical criteria24. The Institutional Ethics Committee of Notre-Dame Hospital approved the use of the human articular tissues, and patients provided signed informed consent.
Chondrocyte culture and treatment
Chondrocytes were released from full-thickness strips of cartilage followed by sequential enzymatic digestion at 37°C, as described25. Cells were seeded at high density (105 cells/cm2) in tissue culture flasks, and cultured to confluence in Dulbecco’s modified Eagle’s medium (DMEM; Wisent, Saint-Bruno, QC, Canada) supplemented with 10% heat-inactivated fetal bovine serum (FBS; PAA Laboratories, Etobicoke, ON, Canada) and an antibiotics mixture (100 units/ml penicillin base and 100 μg/ml streptomycin base; Wisent) at 37°C in a humidified atmosphere of 5% CO2/95% air. To ensure phenotype, we used only first-passage cultured chondrocytes in our study.
At confluence, chondrocytes were treated with GS (Rottapharm, Monza, Italy) at the indicated concentrations for 20 h [for messenger RNA (mRNA) determination] or 72 h (for protein determination) in DMEM containing 0.5% FBS in the presence or absence of IL-1ß 100 pg/ml.
RNA extraction, reverse transcription, and real-time polymerase chain reaction (PCR)
Total cellular RNA from OA chondrocytes was extracted with TRIzol reagent (Invitrogen, Burlington, ON, Canada) according to the manufacturer’s specifications and treated with the DNA-free DNase treatment and removal kit (Ambion, Austin, TX, USA) to ensure complete removal of chromosomal DNA. RNA was quantitated using the RiboGreen RNA quantitation kit (Molecular Probes, Eugene, OR, USA). Reverse transcriptase reactions were primed with random hexamers and real-time quantitation of mRNA was performed as described26 in the Rotor-Gene RG-3000A device (Corbett Research, Mortlake, Australia) with 2X Quantitect SYBRGreen PCR Master Mix (Qiagen, Mississauga, ON, Canada) according to the manufacturer’s specifications. The primer sequences were 5′-GAG TAC TGG AAG CCG AGC AC (sense), 5′-AGG GAC AGG TCT TGG TGT TG (antisense; COX-1); 5′-TGT GTT GAC ATC CAG ATC AC (sense), 5′-ACA TCA TGT TTG AGC CCT GG (antisense; COX-2); 5′-GAA GAA GGC CTT TGC CAA C (sense), 5′-GAA AGG AGT AGA CGA AGC C (antisense; mPGES-1); 5′-TTC TAA AGA GCC TGC GAA AG (sense), 5′-GCA AAC AGC TGT GAG GAC TC (antisense; PPARγ); and 5′-GCA CCA CGT CCA ATG ACA T (sense) and 5′-GTG CGG CTG CTT CCA TAA (antisense). RNA polymerase II (RPII) was used as housekeeping gene.
The primer efficiency for the test gene was the same as for the RPII gene. The data were given as a threshold cycle (CT) and fold changes in gene expression were calculated as 2−ΔΔCT CT over the housekeeping gene RPII. Data are expressed as arbitrary units over control, which was assigned a value of 1.
Western blot examination
Total proteins were extracted with RIPA buffer (Tris-HCl 50 mM, pH 7.4, NP-40 1%; Na-deoxycholate 0.25%, NaCl 150 mM, EDTA 1 mM, and Na-orthovanadate 1 mM) supplemented with protease inhibitors27. The protein level was determined using the bicinchoninic acid protein assay, and 10 μg of the protein was electrophoresed onto a NuPAGE Novex 4-12% Bis-Tris gel. The proteins were transferred electrophoretically onto a nitrocellulose membrane for 1 h at 4°C. The efficiency of transfer was controlled by a brief staining of the membrane with Ponceau Red and destaining in water and TTBS 1× (Tris 20 mM, NaCl 150 mM, pH 7.5, and 0.1% Tween 20) before immunoblotting.
The membranes were incubated overnight at 4°C with 5% skim milk in SuperBlock blocking buffer-blotting in Tris-buffered saline. The membranes were then washed once with TTBS for 10 min and incubated in TTBS 1× with 0.5% skim milk supplemented with the following antibodies: a mouse monoclonal anti-human COX-1 (1:5000), a mouse monoclonal anti-human COX-2 (1:5000), a rabbit polyclonal anti-human mPGES-1 (1:2000; all from Cayman Chemical, Ann Arbor, MI, USA), and a rabbit anti-human GAPDH (1:50,000; Abcam, Cambridge, MA, USA) overnight at 4°C. The membranes were washed with TTBS 1× and incubated 1 h at room temperature with the second antibody (COX-1 and COX-2, 1:20,000 anti-mouse; mPGES-1, 1:10,000 anti-rabbit; GAPDH, 1:50,000 horseradish peroxidase-conjugated anti-rabbit IgG; Pierce Chemical, Rockford, IL, USA) and washed again with TTBS 1×. Detection was performed by chemiluminescence using the Super Signal West Dura extended duration substrate (Pierce) and exposure to Kodak Biomax photographic film. Band intensity was measured by densitometry using TotalLab TL 100 software and data were expressed as arbitrary units of the ratio of the target protein/GAPDH relative to the control, which was assigned the value of 1.
Other determinations
The level of PGE2 was determined in the culture medium and quantified by a specific enzyme immunoassay and that of the glutathione was determined in the cell lysate and quantified by an assay kit (both from Cayman Chemical). All determinations were performed in duplicate for each cell culture.
Statistical analysis
Data are expressed as the mean ± SEM of independent specimens. Statistical significance was assessed by Student’s unpaired or paired t test when appropriate.
RESULTS
Effect of GS on levels of PGE2 production in OA chondrocytes
To determine whether GS plays a role in the PGE2 production in cartilage during the OA process, we investigated the effect of GS on this factor, and further, at which level it acted in the PGE2 pathway. Data showed that in OA chondrocytes, the basal PGE2 production was low and that cells stimulated with IL-1ß exhibited a significant increase (p ≤ 0.0001). Interestingly, in both the absence and presence of IL-1ß, GS dose-dependently inhibited PGE2 production, and significance was reached at 0.2 mM for the endogenous and at 1 mM for IL-1ß-induced production of PGE2 (Figure 1).
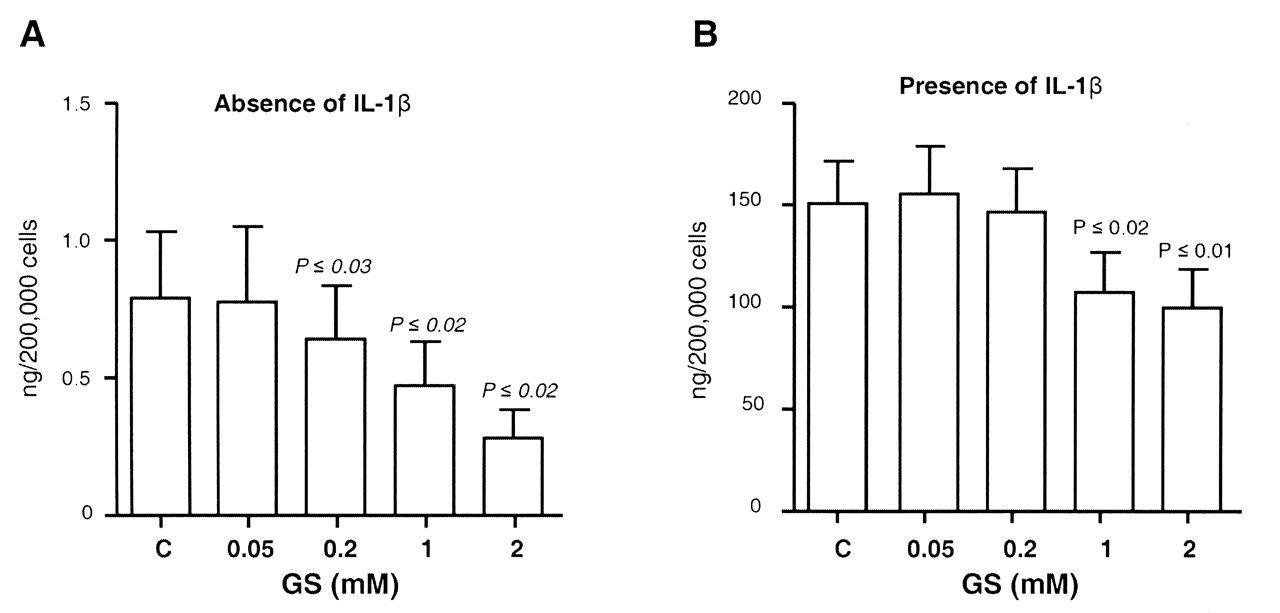
Effect of glucosamine sulfate (GS) on human OA chondrocyte production of prostaglandin E2 (PGE2) in (A) the absence (n = 6) and (B) presence (n = 7–10) of interleukin 1ß (IL-1ß; 100 ng/ml). Cells were treated for 72 h in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period, the culture medium was removed and processed for PGE2 determination. Data are expressed as mean ± SEM and statistical significance was assessed by Student’s t test; p values are versus the control group. Of note, statistical significance was achieved (p ≤ 0.0001) between the control in the absence of IL-1ß and the control in the presence of IL-1ß.
Effect of GS on expression of COX-1, COX-2, and mPGES-1 in OA chondrocytes
Since COX-1, COX-2, and mPGES-1 are the major enzymes involved in production of PGE2, we next examined at which level the effect of GS was attributable. We investigated the effect of GS on the expression pattern of COX-1, COX-2, mPGES-1, and related factors in OA chondrocytes.
Data first revealed that COX-1 mRNA expression was decreased by about 67% (p ≤ 0.0001) by IL-1ß in OA chondrocytes. GS had no effect on COX-1 mRNA expression in either the absence or presence of IL-1ß (Figure 2A, 2B). At the protein level, although significance was not reached, a numerical decrease in the level of COX-1 production under both endogenous and IL-1ß-stimulated cell conditions was observed with GS at a concentration of 2 mM (Figure 2C, 2D).

Effect of glucosamine sulfate (GS) on human osteoarthritis chondrocyte (A, B) gene expression of COX-1 (n = 6) and (C, D) protein level (n = 5) as assessed by Western blot in the absence (A, C) and presence (B, D) of IL-1ß (100 ng/ml). Cells were treated for (A, B) 18 h or (C, D) 72 h in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period (A, B) total RNA was extracted and processed for real-time polymerase chain reaction or (C, D) cells were released and processed for Western blot. Data were calculated over the housekeeping gene RNA polymerase II or protein GAPDH and expressed as mean ± SEM of arbitrary unit over the control, which was attributed a value of 1. Statistical significance was assessed by Student’s t test. mRNA: messenger RNA; IL: interleukin.
As expected, the level of COX-2 expression was significantly stimulated by IL-1ß (15-fold; p ≤ 0.05), and GS did not significantly affect its mRNA level (Figure 3A, 3B). As for protein production, data showed that at lower concentrations the molecular mass of COX-2 is 72–74 kDa. However, at 2 mM in the absence of IL-1ß and at 1 and 2 mM in the presence of IL-1ß, the apparent molecular mass decreased to 66–70 kDa, and in the presence of IL-1ß at 1 and 2 mM, there was a strong accumulation of COX-2 (Figure 3C, 3D). These data are in accord with findings reported for G.HCl in other human cell types28, and indicate that in human OA chondrocytes, GS prevents COX-2 cotranslational N-glycosylation.

Effect of glucosamine sulfate (GS) on human osteoarthritis chondrocyte (A, B) gene expression of COX-2 (n = 6) and (C, D) protein level (n = 6) as assessed by Western blot in the absence (A, C) and presence (B, D) of interleukin 1ß (IL-1ß; 100 ng/ml). Cells were treated for 18 h (A, B) or 72 h (C, D) in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period (A, B), total RNA was extracted and processed for real-time polymerase chain reaction or (C, D) cells were released and processed for Western blot. Data (A, B) were calculated over the housekeeping gene RNA polymerase II and expressed as mean ± SEM of arbitrary unit over the control, which was attributed a value of 1. Statistical significance was assessed by Student’s t test. mRNA: messenger RNA; COX-2: cyclooxygenase-2.
Further experiments were performed downstream of the COX, and we investigated the effect of GS treatment on the expression of mPGES-1, the key terminal synthase responsible for the biosynthesis of the inducible PGE2. As with PGE2, the endogenous expression and production of mPGES-1 were low, and IL-1ß elicited a 5- and 2.3-fold increase (p ≤ 0.05) in levels of mRNA and protein, respectively. GS significantly inhibited the levels of both mPGES-1 mRNA and protein at 2 mM in the absence of IL-1ß and at 1 and 2 mM under IL-1ß stimulation (Figure 4).

Effect of glucosamine sulfate (GS) on human osteoarthritis chondrocyte (A, B) gene expression of mPGES-1 (n = 5–8) and (C, D) protein level (n = 5–8) assessed by Western blot in (A, C) the absence and (B, D) presence of interleukin 1ß (IL-1ß; 100 ng/ml). Cells were treated for (A, B) 18 h or (C, D) 72 h in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period (A, B), total RNA was extracted and processed for real-time polymerase chain reaction or (C, D) cells were released and processed for Western blot. Data were calculated over the housekeeping gene RNA polymerase II or protein GAPDH and expressed as mean ± SEM of arbitrary unit over the control, which was attributed a value of 1. Statistical significance was assessed by Student’s t test. mRNA: messenger RNA.
Effect of GS on glutathione production in OA chondrocytes
It is well established that mPGES-1 requires glutathione as an essential cofactor for its activity. Additional experiments were carried out to verify if the GS-mediated decrease in mPGES-1 production was related to alterations in glutathione production in OA chondrocytes. Data showed that IL-1ß did not modulate glutathione production, but it was modestly yet significantly reduced (p ≤ 0.05) by GS at 2 mM concentration (Figure 5A, 5B).

Effect of glucosamine sulfate (GS) on human osteoarthritis chondrocyte activity of glutathione in (A) the absence (n = 6) and (B) presence (n = 6) of interleukin 1ß (IL-1ß; 100 ng/ml). Cells were treated for 72 h in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period, the cells were released and cell lysates processed for determination of glutathione activity. Data are expressed as mean ± SEM and statistical significance was assessed by Student’s t test; p values are versus the control group.
Effect of GS on PPARγ expression in OA chondrocytes
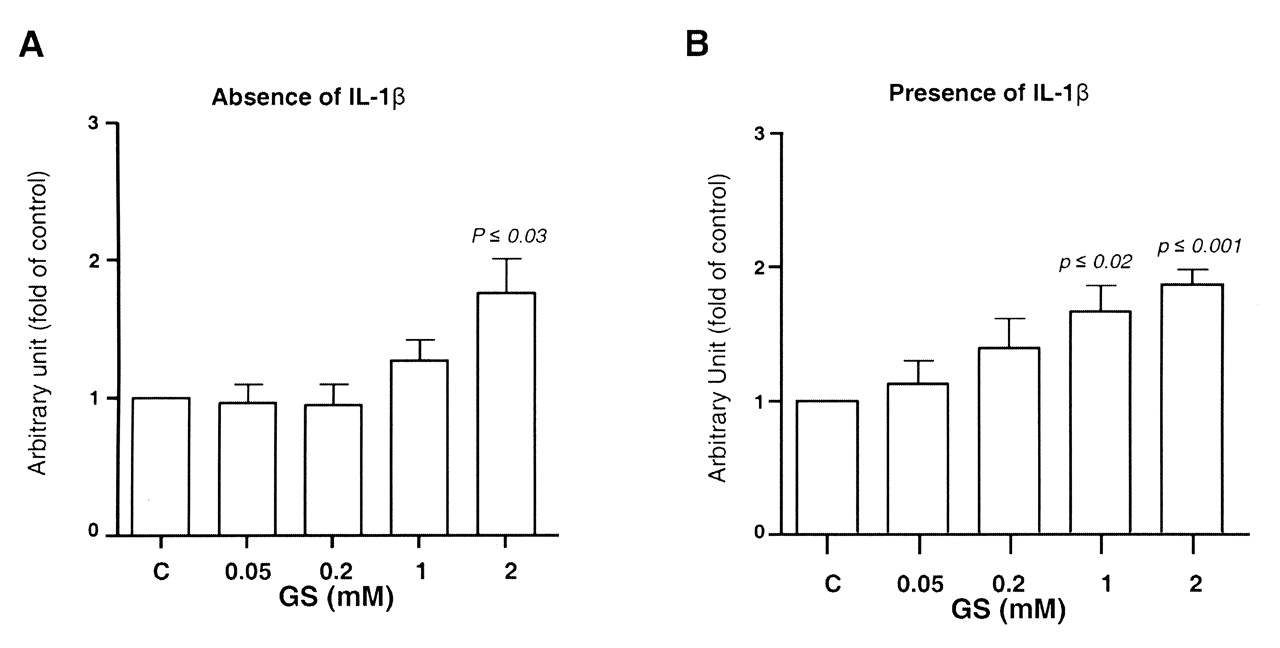
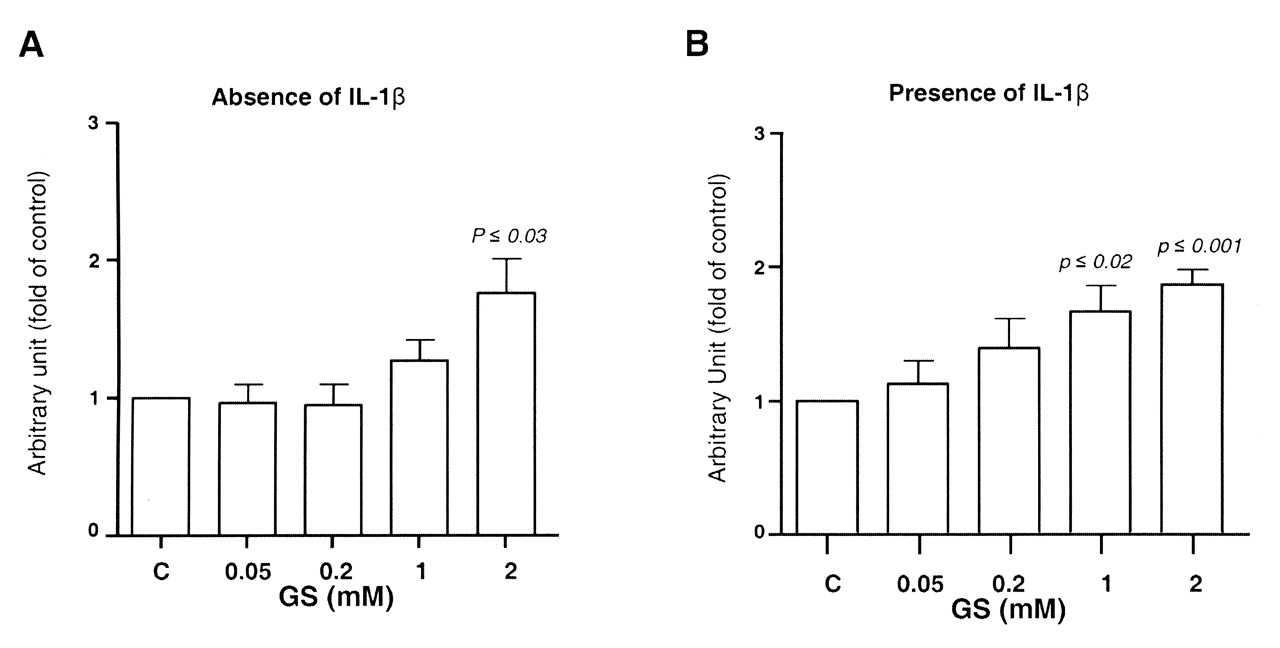
It is also well known that PPARγ activation can inhibit the IL-1ß-mediated induction of mPGES-129. We therefore investigated whether the GS-induced inhibition of mPGES-1 correlated with an increase in the levels of PPARγ. IL-1ß markedly decreased (p ≤ 0.003) PPARγ mRNA expression. As with mPGES-1, in both the absence and presence of IL-1ß, GS induced a significant increase in PPARγ expression in a dose-dependent fashion in OA chondrocytes (Figure 6A, 6B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of glucosamine sulfate (GS) on human osteoarthritis chondrocyte gene expression of peroxisome proliferator-activated receptor-γ and in (A) the absence (n = 6) and (B) presence (n = 6) of interleukin 1ß (IL-1ß; 100 ng/ml). Cells were treated for 18 h in the absence [control (C)] or presence of GS at 0.05, 0.2, 1, and 2 mM. At the end of the incubation period the total RNA was extracted and processed for real-time polymerase chain reaction. Data are expressed as mean ± SEM and statistical significance was assessed by Student’s t test; p values are versus the control group.
DISCUSSION
Increased production of PGE2 is a key event associated with the pathogenesis of OA, and inhibitors of PGE2 biosynthesis are increasingly used for the treatment of this disease and other conditions associated with elevated levels of PGE2. The induced synthesis of PGE2 requires 2 rate-limiting enzymes within the arachidonic acid metabolic pathway: COX-2 and mPGES-1. In our study, GS suppressed IL-1ß-induced PGE2 production that correlates with an inactivation of COX-2 as well as a suppression of the IL-1ß-induced production of mPGES-1 and the activity of its cofactor, glutathione. In addition, GS enhanced the expression of PPARγ, a potent antiinflammatory factor known to inhibit the IL-1ß-mediated induction of mPGES-1.
Glucosamine is a naturally occurring amino monosaccharide that has been shown to exhibit protective properties in OA joint tissues and in other arthritic diseases in humans and animals30. However, the mechanism of action of GS remains unclear.
We observed that GS dose-dependently inhibited IL-1ß-induced PGE2 production but that it was not related to COX-2 mRNA levels. This is in agreement with the findings of Jang, et al28, who demonstrated that GS has no effect on the steady state of COX-2 mRNA or the newly synthesizing COX-2 mRNA in IL-1ß-treated A549 cells. However, this is in contrast to other studies showing that GS suppressed COX-2 expression at the transcriptional level. For instance, Rafi, et al31 reported that G.HCl transcriptionally prevented lipopolysaccharide-induced COX-2 expression by inhibiting NF-κB in murine macrophages. Moreover, Largo, et al showed that treatment with GS suppressed IL-1ß-induced COX-2 expression through transcriptional downregulation and suppression of NF-κB DNA binding activity in human chondrocytes22, and as well, decreased COX-2 expression on peripheral blood mononuclear cells in rabbits with atherosclerosis aggravated by chronic arthritis32. The reasons for these discrepancies are unclear but may relate to differences in the experimental conditions, cell species, and the glucosamine preparation. In this context, Rafi, et al31 used G.HCl, the effect of which may differ from that of GS.
With regard to COX-2 protein our data revealed that IL-1ß induced a strong expression of COX-2 protein, and treatment with GS caused a decrease in the COX-2 protein molecular mass. Interestingly, the appearance of the COX-2 with the low molecular mass coincides with the reduction in PGE2 synthesis. COX-2 is an N-glycoprotein with 4 glycosylation sites33, and it has been shown that inhibition of COX-2 N-glycosylation by site-directed mutagenesis or pharmacologically using an N-glycosylation inhibitor leads to the production of a COX-2 protein with reduced molecular mass and activity34. Therefore, our data suggest that GS suppresses IL-1ß-induced PGE2 production by inhibiting COX-2 N-glycosylation and this factor’s subsequent inactivation. This is supported by recent findings that G.HCl prevented the induction of PGE2 production by IL-1ß, tumor necrosis factor-α, or phorbol 12-myristate 13-acetate through inhibition of COX-2 glycosylation in human skin fibroblasts35, and in the A549 human lung epithelial cells28.
In addition to COX-2, mPGES-1 is a rate-limiting enzyme downstream of COX enzymes and specifically converts prostaglandin H2 to PGE2. mPGES-1 is coupled with COX-2 and catalyzes the terminal step in the biosynthesis of PGE2. We found that GS suppressed both basal and IL-1ß-induced mPGES-1 expression and protein levels. These data thus suggest that GS can suppress PGE2 biosynthesis not only through modulation of COX-2 activity, but also by suppressing mPGES-1 expression. This concurs with data in which treatment with GS suppresses IL-1ß-induced mPGES-1 expression in bovine articular cartilage explants and in equine chondrocytes and synoviocytes21,36,37. Further, our data also showed that GS inhibits the activity of glutathione, an essential cofactor for mPGES-1 activity. Therefore, in addition to its effects on COX-2, GS can also suppress PGE2 biosynthesis by inhibiting mPGES-1 production and its activity.
Several lines of evidence suggest that PPARγ activation may have therapeutic benefits in OA and possibly in other chronic articular diseases. PPARγ activation suppressed the expression of several genes considered essential in the pathogenesis of OA, including IL-1ß, inducible nitric oxide synthase, MMP-1, and MMP-13, and prevented the IL-1ß-induced proteoglycan degradation38,39. PPARγ activation was also shown to inhibit PGE2 production by suppressing the expression of COX-2 and mPGES-129,40. Other studies in animal models of OA demonstrated the protective effect of PPARγ on the diseased articular tissues. Therefore, it was logical to speculate that GS-mediated suppression of PGE2 production could also be regulated by modulation of expression of PPARγ. In that regard, data from our study showed that GS enhanced PPARγ expression in OA chondrocytes, suggesting that it may suppress PGE2 biosynthesis indirectly through upregulation of expression of PPARγ.
A limitation of our study that is inherent to in vitro studies is that the concentrations used for GS may represent supraphysiological therapeutic levels of the drug. The concentrations were chosen according to the most current literature and are within the range of in vitro concentrations used by the majority of investigators. The glucosamine concentration found in plasma and synovial fluid after oral 1500 mg GS in humans is 10 μM. In the literature, such low concentration demonstrated no effect and generally the lowest effective concentration reported was not < 50 μM. However, these experiments were performed on healthy bovine cartilage explants and generally using a long incubation period (14–28 days). Of note, it has been reported that higher concentrations of glucosamine should be used when experiments are performed on OA chondrocytes compared to normal chondrocytes20. Another interesting point is that comparison between experiments done between the culture medium containing 10% serum and serum-free revealed that for the latter condition, glucosamine is much less effective41. This effect was suggested to be related to the presence of factors (e.g., growth factors) in the serum that could contribute to the response. In our current work, experiments were performed with 0.5% serum, which is a minimal concentration generally used to avoid having a biased effect. In addition, previous studies performed with the radiolabeled molecule have shown that glucosamine tends to concentrate in the cartilage, where concentrations may increase with repeated administrations42, and may be much higher than those found in plasma and synovial fluid after administration of therapeutic doses43,44. Of importance also is that the pharmacokinetics of glucosamine is modulated by the levels of glucose in the culture medium, as it uses glucose transporters to be taken up by the cells45,46. Because our cells are grown in a medium with high levels of glucose (DMEM containing 25 mM glucose), it was necessary to use high concentrations of glucosamine in order to appreciate its effect.
In addition, it should be noted that treatment with GS is characterized by a slow onset of action, with a maximal clinical effect being attained after several months, i.e., 3 to 6 months. Hence, in order to reproduce in vitro an effect observed in vivo obtained after several weeks of treatment, it appears to be necessary to increase the drug concentrations for in vitro determination.
Another limitation is that some could argue that in in vitro experiments the GS will dissociate in the culture medium and increase the level of free sulfate concentration, which could influence the results. In fact, it has been shown that the serum level of inorganic sulfate increases after the administration of GS47, and one could hypothesize that it may contribute to the pharmacologic effect of the drug. Such a hypothesis could be supported by recent reports on the in vitro comparison between GS and G.HCl48,49 on cartilage catabolic and anabolic factors showing that both preparations demonstrated similar outcomes, but that GS was more effective. Moreover, although there has not been a head to head clinical study between these preparations of glucosamine (GS and G.HCl), trials showed that GS7,8,9,10,11 in contrast to G.HCl12,50 was effective, in addition to having better pharmacokinetics23.
We have demonstrated that GS inhibits IL-1ß-induced production of PGE2 in human OA chondrocytes, and that the inhibition appears to occur at multiple levels: induction of a shift in the molecular mass of COX-2, and thus its inactivation; inhibition of mPGES-1 expression and production; and augmentation of expression of PPARγ. These findings could at least in part explain the protective effects of GS in OA and other inflammatory arthritides.
Acknowledgment
The authors are grateful to François-Cyril Jolicoeur and Changshan Geng from the Osteoarthritis Research Unit of the CRCHUM for their expert technical assistance, as well as Virginia Wallis for assistance with manuscript preparation.
Footnotes
-
Supported by a grant from Rottapharm, Monza, Italy.
- Accepted for publication September 15, 2011.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
- 49.
- 50.