

Abstract
Objective. To evaluate the safety and efficacy of civamide cream 0.075% for the treatment of osteoarthritis (OA) of the knee.
Methods. We conducted a 12-week, multicenter, randomized, double-blind study with a 52-week open-label extension. Patients with OA of the knee received either civamide cream 0.075% or a lower dose of civamide cream, 0.01%, as the control. The 3 co-primary endpoints in the double-blind study were the time-weighted average (TWA) of change from baseline to Day 84 in the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale, the WOMAC physical function subscale, and the Subject Global Evaluation (SGE). In the 52-week open-label extension study, the Osteoarthritis Pain Score and SGE were assessed.
Results. A total of 695 patients were randomized to receive civamide cream 0.075% (n = 351) or civamide cream 0.01% (control; n = 344) in the double-blind study. Significance in favor of civamide cream 0.075% was achieved for the TWA for all 3 co-primary efficacy variables: WOMAC pain (p = 0.009), WOMAC physical function (p < 0.001), and SGE (p = 0.008); and at Day 84 for these 3 variables (p = 0.013, p < 0.001, and p = 0.049, respectively). These analyses accounted for significant baseline-by-treatment interactions. In the 52-week open-label extension, efficacy was maintained. Civamide cream 0.075% was well tolerated throughout the studies.
Conclusion. These studies demonstrate the efficacy of civamide cream for up to 1 year of continuous use. Civamide cream, with its lack of systemic absorption, does not have the potential for serious systemic toxicity, in contrast to several other OA treatments.
Osteoarthritis (OA), the most common joint disorder, is an active process involving bone, cartilage, ligament, muscle, and synovial joint tissues. Symptoms of OA usually appear in middle or late age and involve deep aching pain, joint crepitus, swelling, stiffness, and limitation in movements of the weight-bearing joints, hands and spine1,2. The majority of patients with OA complain that pain is their worst problem3. OA symptoms are often treated with acetaminophen. Large doses have been associated with hepatic toxicity. Also used are nonsteroidal antiinflammatory drugs (NSAID), salicylates, or cyclooxygenase (COX)-2 inhibitors. These drugs have the potential for significant gastrointestinal (GI; including peptic ulcer disease), cardiovascular, and renal side effects. These drugs may prove insufficient to control OA pain, justifying the use of more potent analgesic agents such as opioids, which can induce impaired GI motility, respiratory depression, dizziness, somnolence, other central nervous system (CNS) side effects, and addiction. The availability of an effective topical medication that is not systemically absorbed, and thus without the risk of either drug-related systemic adverse events or drug-drug interactions, is additionally advantageous for an older population at greater risk for these events.
Civamide (cis-8-methyl-N-vanillyl-6-nonenamide) is a TRPV-1 receptor modulator that acts by a mechanism similar to capsaicinoids such as capsaicin4,5,6,7,8. Capsaicinoids selectively depress type-C nociceptive fibers by causing a calcium ion-dependent release of substance P and subsequent desensitization to its further release9. The result is an attenuation of the conduction and transmission of peripheral pain impulses centrally from the joint.
Systemic absorption of civamide has not been detected in a pharmacokinetics study of civamide cream 0.075% applied to the skin around the knee joint in human patients10. As there is no measurable systemic absorption, there is no potential for systemic side effects such as hepatic, GI, or cardiovascular effects from this therapy, making civamide particularly valuable for patients who do not tolerate or are not adequately treated by oral medications. In previous clinical studies, civamide cream 0.075% was found to improve the signs and symptoms of OA of the knee. It reduced OA pain severity and improved both patient and physician global evaluations11,12.
The primary aim of our studies was to examine the safety and efficacy of civamide cream 0.075% over a 3-month period, and to examine the safety and continued effect for a longer period (up to 1 year), for the treatment of symptomatic knee OA.
MATERIALS AND METHODS
Fifty clinical centers in the United States participated in the 12-week pivotal double-blind study between June 2003 and June 2004. Thirty-three of these clinical centers participated in the 52-week open-label extension study between January 2004 and July 2005. The protocols and written informed consent forms were approved by an institutional review board for each study site. Although both studies were conducted before trial registry was required, the longterm study was registered prior to enrollment of subjects (ClinicalTrials.gov identifier NCT00077935).
Patients
Adults over 50 years of age who had OA for at least 6 months and whose OA diagnosis met the American College of Rheumatology criteria were eligible to participate in the 12-week double-blind study. Eligible patients were to have a Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscale baseline value of ≥ 9 (0–20 scale), Functional Capacity Classification of I–III13, a radiograph and/or report detailing the results within 3 years of screening that showed radiographic evidence of OA of the target knee with Kellgren-Lawrence scale of 2 or 3, and either morning stiffness < 30 min duration or crepitus on active motion present upon examination. Patients were also required to be taking a stable dose of NSAID or COX-2 inhibitors for pain for at least 28 days prior to entry, with no interruption for 2 days prior to the screening and baseline visits.
Patients were excluded from the double-blind study for evidence of other conditions or diseases of the skin or joint and for partial or complete knee joint replacement or anticipated joint replacement of the target knee. Additional exclusion criteria were history or diagnosis of other arthritic conditions, rheumatoid arthritis, fibromyalgia and/or other inflammatory and immune system disorders, diabetes Type I or II, obesity (body mass index ≥ 39), painful conditions or frequent headaches requiring use of systemic opiates or derivatives, or need for additional NSAID or COX-2 inhibitors. Patients with known allergy or sensitivity to capsicum, civamide, or capsaicin-containing products or constituents of the cream formulation were also excluded.
Study design
This was a 12-week, randomized, double-blind, controlled, 2-arm, multicenter evaluation of the safety and efficacy of civamide cream 0.075% and civamide cream 0.01% (control) with a 52-week open-label extension to evaluate the safety of longterm treatment with civamide cream 0.075% (Figure 1). Clinical protocol numbers were assigned to each study (WL-1001-05-01 and WL-1001-05-04, respectively). In order to protect the blinding to treatment assignment, the 12-week study was designed with a control cream containing a low dose (0.01%) of the active ingredient (civamide) that initially produces localized burning sensations similar to those of the 0.075% study drug. The use of civamide cream 0.01% as the control was not a placebo control in that the activity of civamide in the cream in improving the signs and symptoms of OA was recognized as a possibility. A time-weighted average (TWA) analysis of the data of the co-primary efficacy variable was planned to account for this. The dose and dose interval selection were based on previous phase I tolerability studies and phase II studies using civamide cream 0.075% in the treatment of OA.
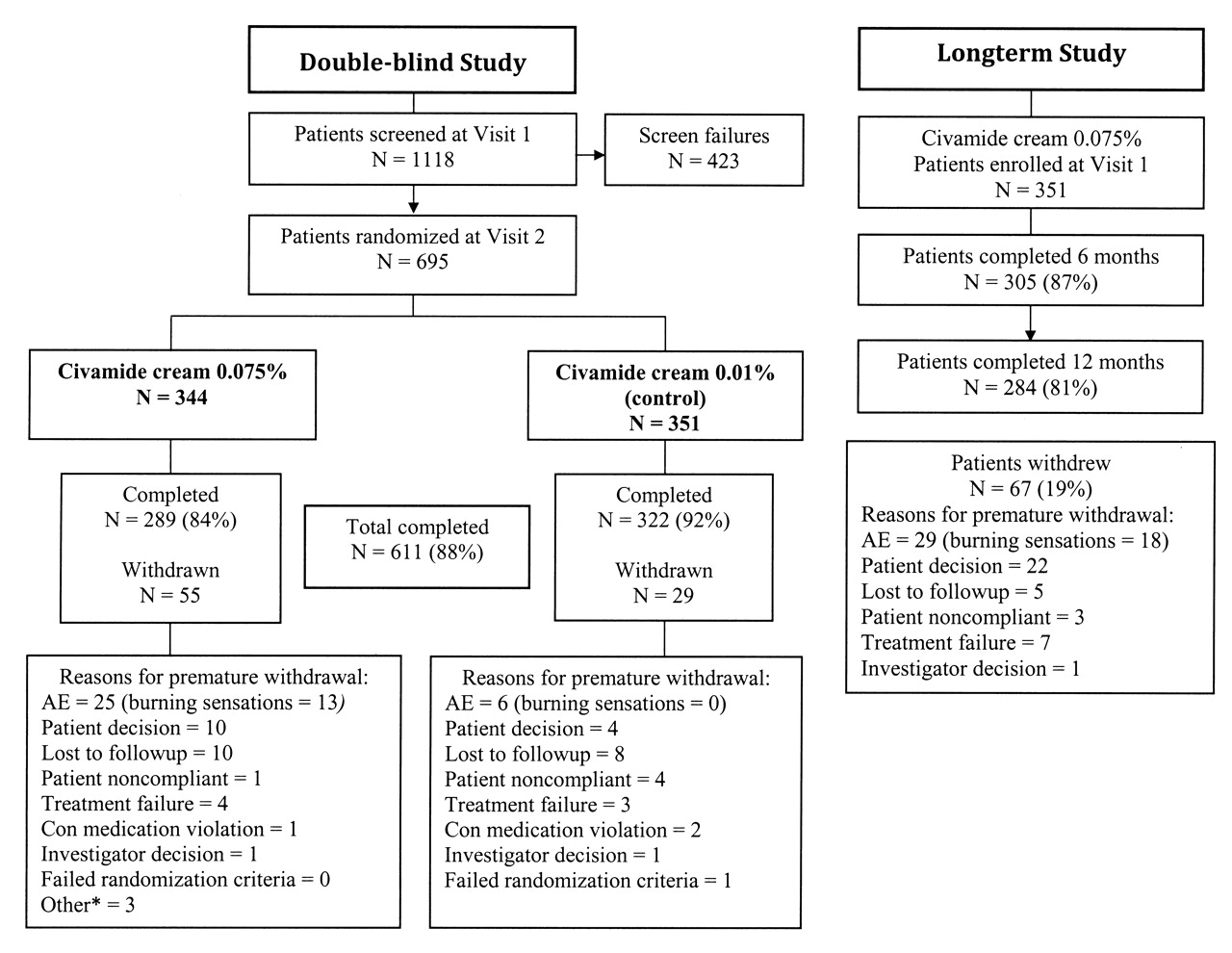
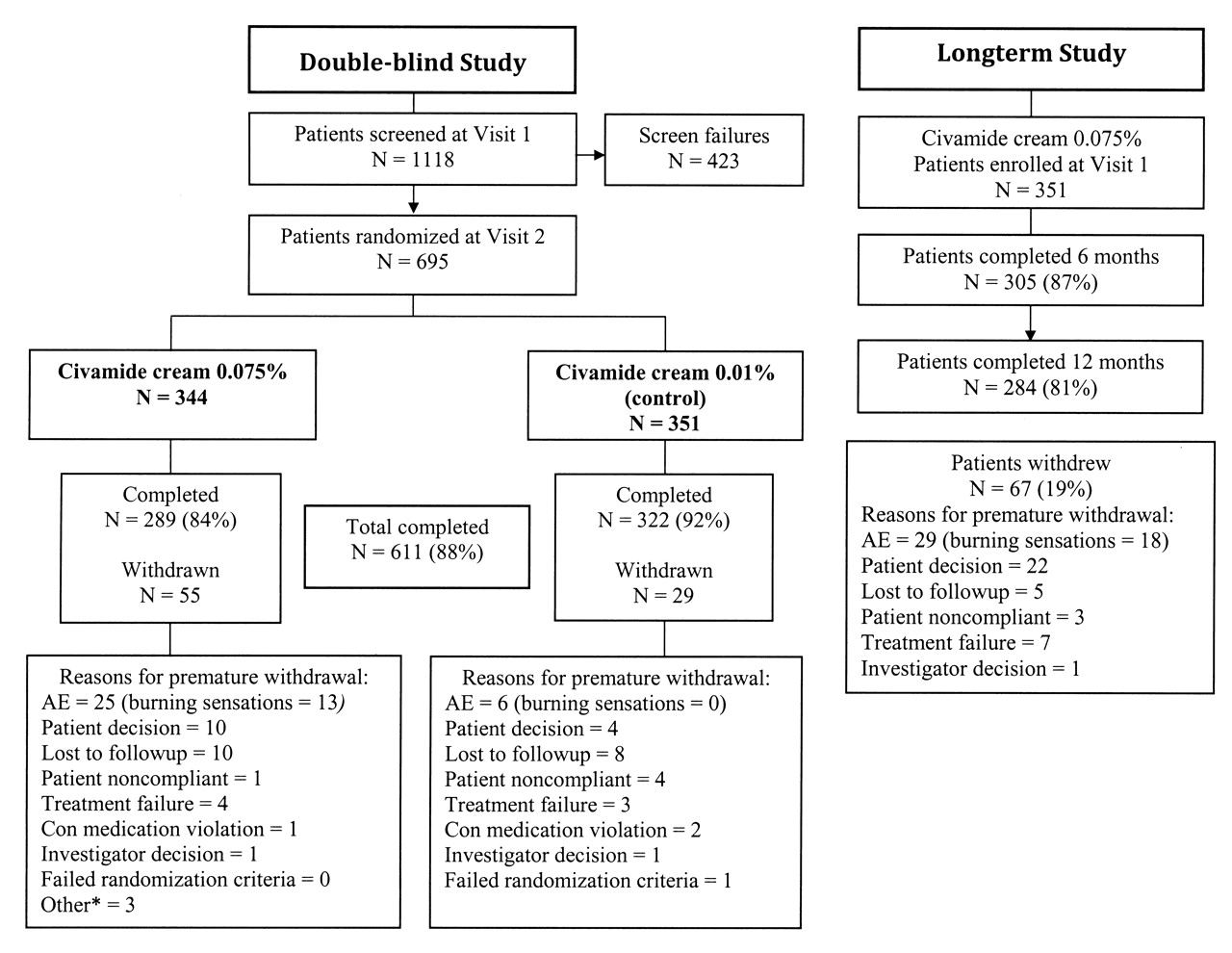
Patient disposition in the double-blind study and in the longterm study. AE: adverse event. *Discontinued for reasons other than those listed.
Eligible patients entered the double-blind treatment period and were subsequently randomized in a 1:1 ratio to active or control cream 3 times daily (TID) for 12 weeks to treat their target knee. Patients were not allowed to treat their other knee. Patients were instructed to apply a small amount of cream (pea size) to each of 3 locations around the target knee, and using 1 or 2 fingers to gently rub the cream in, leaving no residue on the skin. Assessments for efficacy and safety were performed at clinic visits on Days 21, 42, 63, and 84/final visit. During the study, each patient recorded in a daily diary information regarding study drug usage, stable oral OA medication usage, other concurrent medication use, and adverse events. Patients were also contacted by phone on Days 15, 36, 57, and 78 to discuss adherence to therapy, concurrent medication use, and adverse events. Blinding was additionally maintained by designating separate efficacy rater and safety rater staff to perform specific procedures.
Upon completion of the 12-week double-blind study, patients at sites participating in the 52-week longterm extension study were invited to enroll in that study. All enrolled patients treated their target knee TID with civamide cream 0.075%. In that study, patients were also permitted to treat the other knee if affected by OA pain. Assessments for efficacy and safety were performed at clinic visits on Day 1 and Weeks 13, 26, 39, and 52 (end of study/final visit). Patients recorded concurrent medications and adverse events in a diary.
Permitted and excluded medications and treatments
In the double-blind trial, stable use of an NSAID or COX-2 inhibitor medication was required for at least 22 of the 28 days prior to entry without interruption for each of the 2 days prior to the screening visit (Visit 1) and baseline visit (Visit 2). Stable doses of other medications such as systemic antidepressants, anxiolytics, sedative-hypnotics, aspirin ≤ 325 mg daily for cardioprophylaxis, oral contraceptives, estrogen, glucosamine, and nutraceuticals were required for 30–90 days prior to enrollment. Medications that were not permitted included capsaicin and related products, acetaminophen, tramadol, topical medications, creams, ointments, lotions and patches, skeletal muscle relaxants, systemic opiates or derivatives, corticosteroids, injections of hyaluronic acid to the knee, and other investigational drugs. Each of the excluded medications required a washout period of 48 hours to 6 months prior to entry.
For the 52-week study, capsaicin products and other topical products for the treatment area were not permitted, but there were no restrictions or requirements for oral medications. Treatments such as transcutaneous electrical nerve stimulation and invasive procedures affecting the treatment area were not permitted throughout either study.
Efficacy assessments
For the double-blind study, the 3 co-primary efficacy variables were the TWA of the change from baseline (area under the curve of changes divided by total days of observation) during the treatment period of the target knee for the WOMAC pain subscale, for the WOMAC physical function subscale, and for the Subject Global Evaluation (SGE). The use of the TWA analysis as the primary approach was specified in the protocol and agreed upon with the US Food and Drug Administration for the purpose of demonstrating the totality of relief over the entire treatment period. The TWA analysis is viewed to be a more relevant indicator of clinical effect than an analysis at a single timepoint.
The WOMAC Osteoarthritis Index14 is a tridimensional, self-administered, subject-oriented health status questionnaire that was completed at every visit. It consists of 24 items divided into 3 categories: (1) pain subscale with 5 items, (2) stiffness subscale with 2 items, and (3) physical function subscale with 17 items. For this study, each item was scored using a 5-point Likert scale: 0 = none, 1 = slight, 2 = moderate, 3 = severe, and 4 = extreme. The index was calculated as the sum of the 3 subscales and can range from 0 to 96. The SGE is a self-administered scale and was completed at every visit. Based on the OA signs and symptoms of the target knee within the past 24 hours, patients provided a global evaluation using a 5-point Likert scale: 0 = very poor, 1 = poor, 2 = fair, 3 = good, and 4 = very good.
The key secondary efficacy endpoints included the time-specific numeric changes from baseline to Days 21, 42, 63, and 84 for the WOMAC pain subscale, the WOMAC physical function subscale, and the SGE, as well as the total WOMAC OA index score, the WOMAC joint stiffness subscale, the Functional Capacity Classification, and the Medical Outcomes Study Short-Form 36 (SF-36v2) Health Survey. The Functional Capacity Classification, completed at screening and Day 84, uses a 4-point Likert scale and was evaluated by the rater physician across 4 class categories (Class I–IV) ranging from complete functional capacity to incapacitation13. The SF-36v215 is a 4-week recall version (standard form) of the generic, multi-purpose, short-form health survey with 36 questions graded on Likert scales with 3 to 6 possible answers and was completed at baseline and Day 84.
The Outcome Measures in Rheumatology Clinical Trials-Osteoarthritis Research Society International (OMERACT-OARSI) Responder Index was calculated for both treatment groups. This responder analysis is considered to be a validated and useful assessment of clinical response in studies of OA medications and is used internationally16. The OMERACT-OARSI has significant weight in terms of outcome measures for clinical response to treatment in OA trials to assess the comparative clinical response of 2 agents by distinguishing between responders and nonresponders. To be considered a responder, i.e., to have a positive OARSI Simplified Response, the patient had to meet at least 1 of these response conditions: (1) for High Improvement, at least 50% and at least 20 mm on the 100 mm visual analog scale (VAS) for either pain or function; or (2) for Improvement, the occurrence of at least 2 of the following: (a) a decrease in pain of at least 20% and at least 10 mm on the 100 mm VAS; (b) an improvement in function of at least 20% and a decrease of at least 10 mm on the 100 mm VAS; or (c) an improvement in the patient’s global assessment score by at least 20% and at least 10 mm on the 100 mm VAS.
In our study, because the VAS was not measured, the OARSI Simplified Response criteria were assessed by translating the requirements of High Improvement and Improvement by using a linear translation of the VAS scale to the WOMAC pain subscale, the WOMAC physical function subscale, and the SGE. Thus, for example, the High Improvement criteria of 50% change and at least 20 mm on the 100 mm VAS for pain translated, respectively, into an improvement of at least 50% and a decrease of at least 4 units on the WOMAC pain subscale score. The WOMAC Index has been validated for use in both the Likert-scaled version and the VAS-scaled version and the scoring manual for each version supports the linear translation of scores that we performed14.
For the longterm study, the efficacy variables were the change from baseline to each clinic visit of the target knee for the OA pain score and the SGE. Based on OA pain in the target knee within the past 24 hours, patients provided an evaluation using an 11-point numeric rating scale ranging from 0 (no pain) to 10 (worst possible pain).
Safety assessments
Safety assessment for both the double-blind and longterm studies was based on adverse events as well as reported changes from baseline to the end of treatment for physical examination, vital signs, and laboratory tests. Clinically significant changes, as identified by the investigator, in these measurements were recorded as adverse events. Adverse events were coded using MedDRA (Version 5.0, MedDRA MSSO, Reston, VA, USA) for purposes of summary by system/organ class and preferred term. Concurrent medications were coded using the World Health Organization (WHO)-Anatomical Therapeutic Chemical 4 coding system (WHO Collaborating Centre for Drug Statistics Methodology, Norwegian Institute of Public Health, Oslo, Norway).
Statistical analyses
The objective of the double-blind study was to compare the safety and efficacy of civamide cream 0.075% to the control, civamide cream 0.01%. There were 3 co-primary efficacy variables, each involving the TWA of change from baseline: the WOMAC pain subscale, the WOMAC physical function subscale, and the SGE. In accord with the International Conference on Harmonization E917 on the Statistical Principles for Clinical Trials, the protocol for this study specified that p ≤ 0.05 must be met for each of the co-primary variables on the intention-to-treat (ITT) population to demonstrate efficacy. Hence, no adjustment of p values was required for the 3 co-primary variables.
The protocol specified that the 3 co-primary variables as well as the secondary variables, consisting of time-specific numeric change at Days 21, 42, 63, and 84 for WOMAC pain, WOMAC physical function, and SGE, were to be analyzed using a general linear model with treatment and center (pooled sites) as factors and the appropriate baseline variable as a covariate. The protocol also specified that to appropriately use this model, baseline-by-treatment interactions needed to be checked. Further, the protocol specified that if these interactions were visually apparent and statistically significant, assessment of efficacy would use suitable models accounting for the nature of the interactions. These analyses are based on the ITT population and are used last-observation-carried-forward to handle missing data.
Statistically significant baseline-by-treatment interactions were detected in the analysis and visualization, including Loess smoothed curves, of each of the 3 co-primary variables and the important secondary variables of change from baseline to Day 84 for the same 3 variables. As a result, models based on these graphical and analytic results were developed to assess treatment effects. In particular, the visualizations and modeling showed that for low values of the corresponding baseline variables for pain and function, it appeared not possible to distinguish a difference between the 2 treatments, but that for values above a particular baseline value, termed a breakpoint value, the 2 treatments differed. Thus, the models used in the analyses for WOMAC pain and WOMAC physical function used piecewise linear models that assumed no treatment differences for baseline values below the breakpoints and assessed the difference between treatments for baseline values larger than the breakpoint. The breakpoints were chosen analytically through standard statistical model selection by picking the value that minimized the sums of squared errors. For the SGE, due to its discrete scale, an alternative analytic model used Bonferroni adjusted p values from standard linear regressions. For the analyses at Days 21, 42, 63, and 84, models paralleling those of the corresponding co-primary variables were respectively used.
The proportions of patients reporting a treatment-emergent adverse event (TEAE), that is, one beginning or worsening during treatment with the study drug, were compared between treatment groups using Fisher’s exact test. The difference between treatment groups for numeric laboratory tests was assessed by 2-sample t tests on change from baseline, and paired t tests were used to assess within-group changes. The differences between treatment groups for vital signs were also assessed by t tests on change from baseline. New physical examination findings that were deemed clinically significant by the investigator were recorded as adverse events. Adherence to therapy was summarized by treatment group. The percentage of patients reporting concurrent medication usage was summarized. The Fisher’s exact test, the 2-sample t tests, and the paired t tests were all specified.
Although the OMERACT-OARSI response criteria had not been defined at the time the study was designed, this important responder analysis is presented here. In light of the significant baseline-by-treatment interaction that was observed for the WOMAC pain subscale, we also investigated the OMERACT-OARSI response in groups of patients with baseline WOMAC pain subscale scores > 10 and baseline WOMAC pain subscale scores > 13. Baseline values > 13 on the WOMAC pain subscale represent scores between 70% and 100% of the pain scale used. This corresponds to 70 mm to 100 mm on the 100 mm VAS or 7 to 10 on an 11-point numerical rating scale (NRS). Patients with baseline WOMAC pain subscale scores > 13 were considered to have severe pain. This categorization is supported by a review of published literature on pain scales. A verbal rating score (VRS) of “severe” has been shown in several studies to correspond to a value of 70%–100% of either a VAS or NRS scale18. The VRS of “severe” has also been reported to correspond to the mean value of a VAS of 75 mm19, and in another publication, on a 10-point scale (“little” to “terrible”), a VRS of “severe” corresponded to mean VAS scores of at least 73.6 mm20.
The primary objective of the longterm study was to gather longterm safety data of civamide cream 0.075%, which all patients used. The incidence of TEAE was summarized and tabulated. A summary of the number and percentage of patients taking OA medication was provided at each visit. Change from longterm baseline to longterm final visit in each laboratory measure was summarized and the significance of changes was assessed using paired t tests. Summaries of the actual values of systolic blood pressure, diastolic blood pressure, heart rate, and respiration rate were provided for each study visit for all patients. Missing data were not imputed. Any change from baseline in physical examination finding or in a vital sign deemed clinically significant by the investigator was to be recorded as an adverse event. Change from the longterm baseline OA pain score and SGE rating were analyzed for the ITT population at each of the longterm visits at 3, 6, 9, and 12 months, using the paired t test.
RESULTS
Patients
Of 1118 patients screened, 695 patients were enrolled and randomized into the double-blind study and treated at 50 sites (Figure 1). A total of 611 patients (88%) completed the double-blind study. Thirty-three of those sites participated in the longterm safety study, enrolling 351 patients of the potentially 528 patients enrolled in the double-blind study at those sites. Of those 351 patients, 163 (46%) received civamide cream 0.075% and 188 (54%) received civamide cream 0.01% (control) during the double-blind study. One hundred seventy-eight patients entered the longterm study ≤ 30 days after completing the double-blind study and 173 patients at > 30 days. Two hundred eighty-four patients (81%) completed the longterm study.
Baseline patient demographic data for the double-blind and longterm studies are summarized in Table 1. All demographics were comparable between the 2 groups in the double-blind study. Demographics for the longterm study were similar to the double-blind study.
Baseline patient characteristics for the double-blind study and longterm study intention-to-treat population.
Premature withdrawal due to adverse event
In the double-blind study, a total of 32 patients (5%) reported an adverse event that led to premature withdrawal (Table 2). Twenty-five (7%) of the patients in the civamide cream 0.075% group compared to 7 (2%) in the civamide cream 0.01% (control) group withdrew prematurely due to an adverse event. For adverse events related to treatment, 16 patients (5%) who used civamide cream 0.075% and 1 patient (0%) who used civamide cream 0.01% withdrew. Of the 16 civamide cream 0.075% patients who withdrew for an adverse event related to treatment, 13 (4%) reported burning, 1 (< 1%) reported a rash, 1 (< 1%) reported congestion, and 1 (< 1%) reported coughing as the reason for withdrawal. The 1 civamide cream 0.01% patient (< 1%) who withdrew for an adverse event related to treatment reported rash as the reason for withdrawal.
Patient withdrawals* from the double-blind study and the longterm study.
In the longterm study, 29 patients (8%) reported adverse events that led to premature withdrawal (Table 2). Of these, 20 patients (6%) were withdrawn for adverse experiences related to treatment, of which 18 (5%) reported burning, 1 (< 1%) reported friction from pant leg causing knee discomfort, and 1 (< 1%) reported perineal irritation as the reason for withdrawal.
Table 2 displays the premature withdrawals for any reason for both studies.
Efficacy Double-blind study. Pain
The estimated mean responses based on the statistical model for the WOMAC pain subscale TWA are depicted in Figure 2. Civamide cream 0.075% was demonstrated to be significantly more efficacious than civamide cream 0.01% (control) treatment for the co-primary endpoint TWA of change from baseline to Day 84 in WOMAC pain subscale (p = 0.009). The p value corresponds to treatment effects for patients with baseline scores > 10. As WOMAC pain baseline scores increase above 10, civamide cream 0.075% was even more efficacious than civamide cream 0.01% (control).
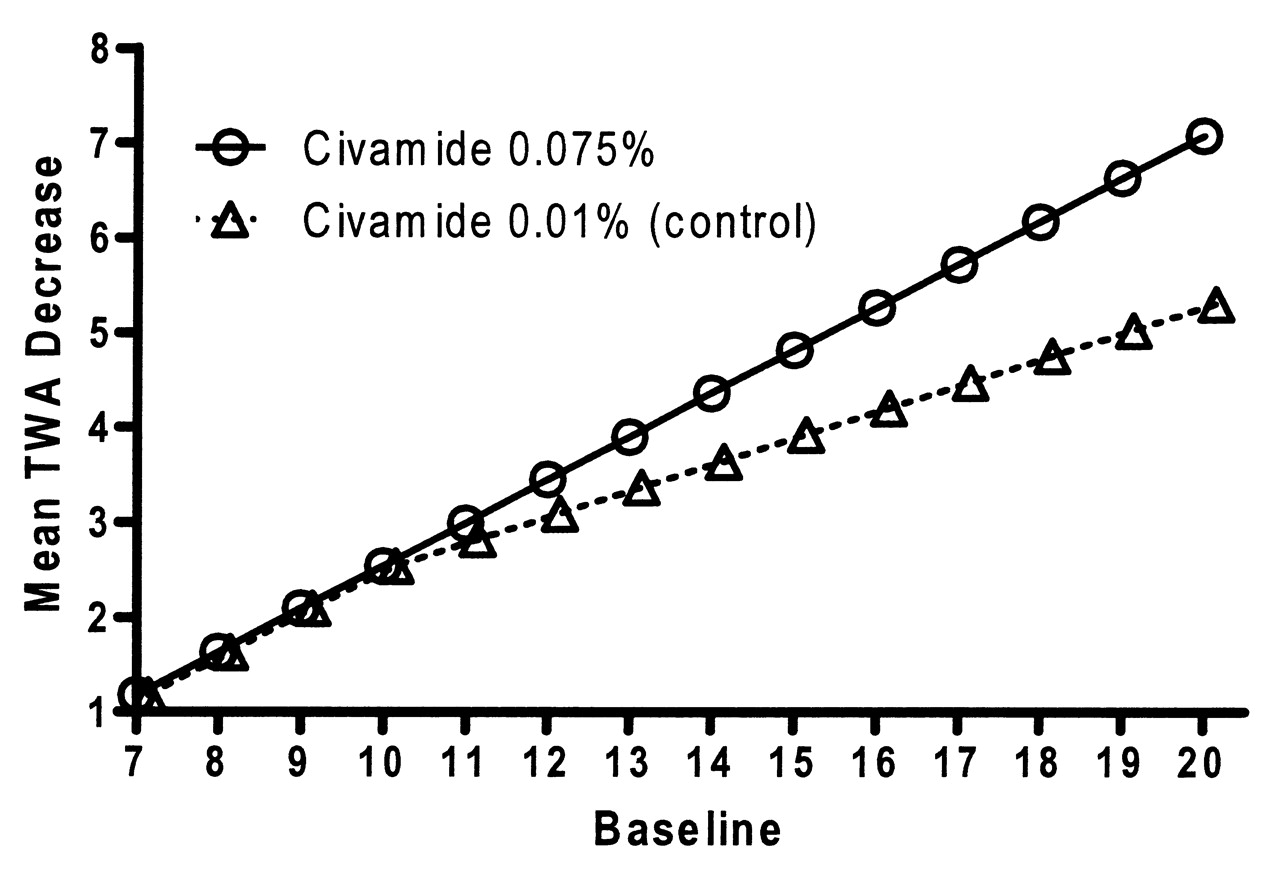
Mean time-weighted average (TWA) from baseline in Western Ontario and McMaster Universities Osteoarthritis Index pain subscale for the double-blind study (intention-to-treat population based on the piecewise linear regression model).
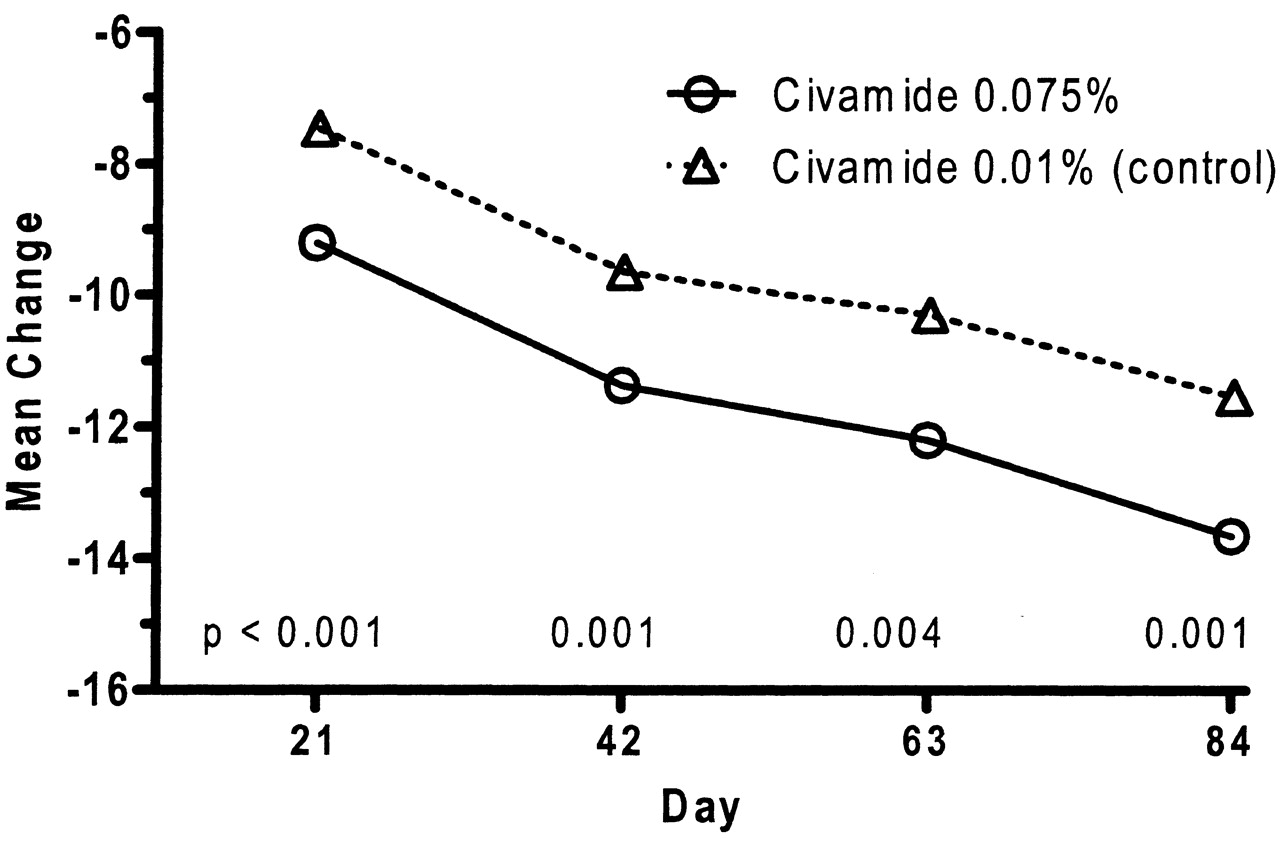
For the time-specific numeric change from baseline of the WOMAC pain subscale, statistical significance was achieved at timepoints Day 21, Day 42, and Day 84, as well as having a p value of 0.0503 at Day 63 (p values for patients with baseline scores > 10). Figure 3 shows the plots based on all ITT patients of the observed mean changes from baseline at Days 21, 42, 63, and 84 for the WOMAC pain subscale. The separation of the means between the civamide cream 0.075% and civamide cream 0.01% (control) generally continues to increase throughout the study.

Numeric change from baseline in Western Ontario and McMaster Universities Osteoarthritis Index pain subscale for the double-blind study intention-to-treat population. P values correspond to treatment differences for baseline scores > 10 (change equals value minus baseline).
Statistically significant results (p = 0.006) were also seen in the OMERACT-OARSI for the ITT population, with a clinically relevant treatment effect of 10%. The treatment effect was 14% (p = 0.002) in patients with baseline WOMAC pain subscale scores > 10 and was even greater in patients with baseline WOMAC pain subscale scores > 13, i.e., with severe pain, with the treatment effect increasing to 27% (p < 0.001; Table 3).
Osteoarthritis Research Society International (OARSI) Simplified Response in intention-to-treat (ITT) population and patients with baseline Western Ontario and McMaster Universities Osteoarthritis Index pain subscale scores > 10 and > 13 for the double-blind study.
Physical function
The estimated mean responses based upon the statistical model for WOMAC physical function subscale are depicted in Figure 4. Civamide cream 0.075% was demonstrated to be significantly more efficacious than civamide cream 0.01% (control) treatment for the co-primary endpoint TWA of change from baseline to Day 84 in the WOMAC physical function subscale (p < 0.001). The p value corresponds to treatment effects for patients with baseline scores > 39.
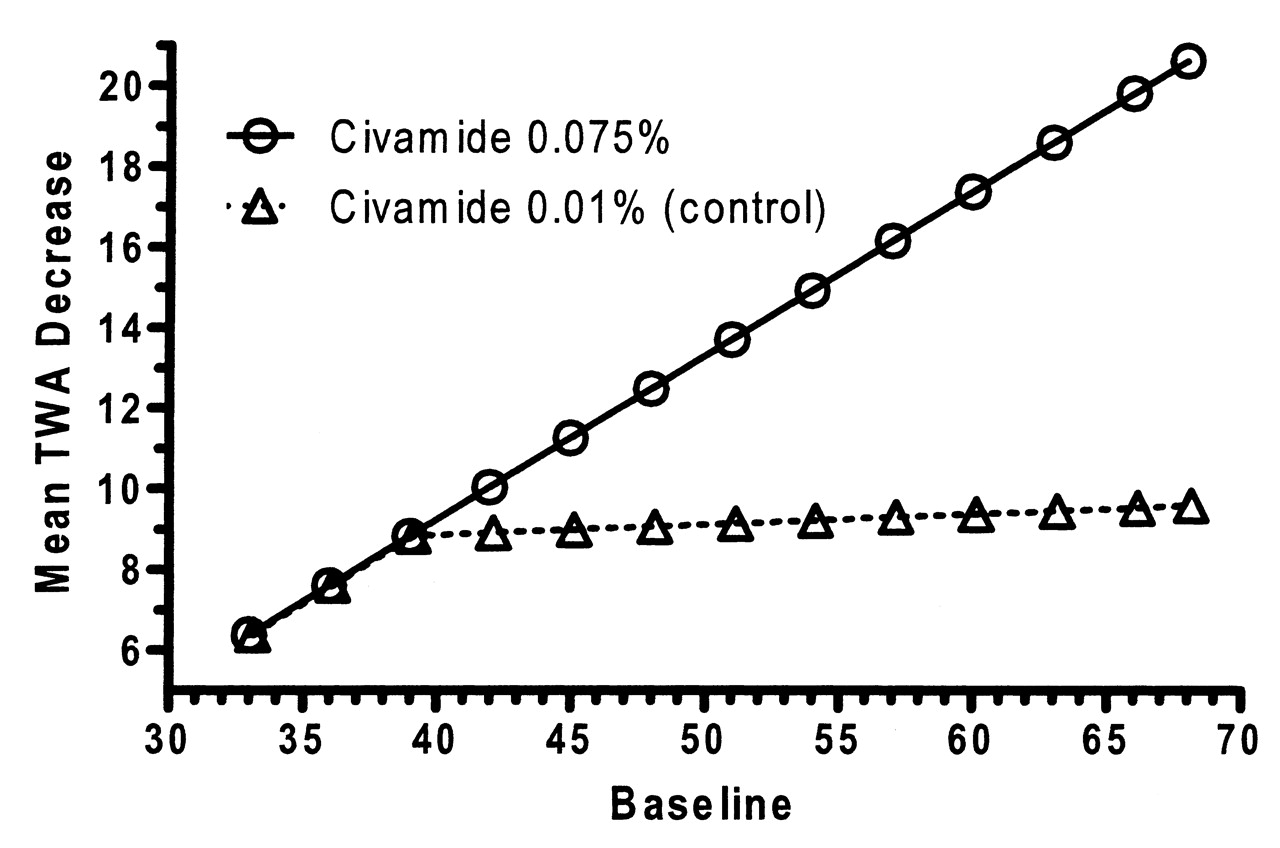
Mean time-weighted average (TWA) change from baseline in Western Ontario and McMaster Universities Osteoarthritis Index physical function subscale for the double-blind study (intention-to-treat population based on the piecewise linear regression model).
As with the WOMAC pain subscale, the data showed that for the WOMAC physical function subscale, civamide cream 0.075% has increasing efficacy compared to civamide cream 0.01% (control) in those patients with increasing baseline scores above 39.
For the time-specific numeric change from baseline of the WOMAC physical function subscale, statistical significance was achieved at all 4 timepoints for patients with baseline WOMAC physical function scores > 39. Figure 5 shows the plots of the observed mean changes from baseline at Days 21, 42, 63, and 84 for the WOMAC physical function subscale.
Numeric change from baseline in Western Ontario and McMaster Universities Osteoarthritis Index physical function subscale for the double-blind study intention-to-treat population. P values correspond to treatment differences for baseline scores > 39 (change equals value minus baseline).
Subject Global Evaluation
For the SGE, statistical significance between the civamide cream 0.075% and civamide cream 0.01% (control) was achieved for the co-primary endpoint TWA of change from baseline to Day 84 (p = 0.008; p value corresponds to treatment effects for patients with baseline scores ≤ 1, i.e., the more severe scores). For the time-specific numeric change from baseline, significance was achieved at 2 of the 4 timepoints, Day 42 (p = 0.009) and Day 84 (p = 0.049; p values correspond to treatment differences for baseline scores = 0).
Secondary endpoints
There was consistency in the secondary endpoints of the double-blind study in support of the benefit of civamide cream 0.075% over civamide cream 0.01% (control) based on the Total WOMAC Index score, the WOMAC stiffness subscale, the Functional Capacity Classification (FCC), and the SF-36v2. In all cases, except for some components in the SF-36v2, observed improvement in the civamide cream 0.075% group was greater than that in the civamide cream 0.01% (control) group, and in many cases civamide cream 0.075% was statistically significantly superior to control.
At Day 84, civamide cream 0.075% was statistically significantly superior to civamide cream 0.01% (control) for the total WOMAC Index score (p = 0.002, corresponding to treatment differences for baseline scores > 60, of total 96) and the WOMAC stiffness subscale (p < 0.05, corresponding to treatment differences for baseline scores > 6, i.e., the more severe scores) at Day 84.
For the FCC, the average changes increased from Day 1 to Day 84 in both groups, with greater change seen in the civamide cream 0.075% group, but the difference between groups was not statistically significant. Similarly, for the SF-36v2, there were no statistically significant differences seen between groups for any component.
Sensitivity analyses
Sensitivity analyses were done using alternative forms of imputation to confirm the robustness of the primary efficacy results: baseline observation carried forward (BOCF) and worst observation carried forward (WOCF) on the change from baseline to Day 84. The p values corresponding to treatment differences are as follows: (1) for baseline WOMAC pain scores > 10 (p = 0.003 for BOCF and p < 0.001 for WOCF); (2) for WOMAC physical function scores > 39 (p < 0.001 for BOCF and p < 0.001 for WOCF); and (3) for baseline SGE scores of 0 (p = 0.033 for BOCF and p > 0.05 for WOCF, both reflecting Bonferroni adjustments). These analyses also take into account the significant baseline-by-treatment interactions.
Longterm study
In the longterm study, patients treated with civamide cream 0.075% had a 26% decrease in pain from the longterm baseline OA pain score as measured in the target knee at 3 months, 30% at 6 months, 30% at 9 months, and continued improvement to a 34% decrease in pain from baseline at 12 months (p < 0.001 at each timepoint by paired t test). Patients treated with civamide cream 0.075% had a 35% improvement from baseline in SGE of the target knee at 3 months, which was maintained at each visit during the study (p < 0.001 at each timepoint by paired t test).
In the longterm study, analysis of patients who either stopped or reduced oral NSAID or COX-2 during the year-long study showed that of the 267 patients who began the study taking an oral pain medication, 60 patients (23%) either discontinued or reduced use of OA medications.
Safety
In the double-blind study, the safety population included all randomized patients who applied at least 1 dose of randomized study drug and had any post-application safety data. Patients in both groups were adherent to the TID study drug dosing regimen: the civamide cream 0.075% group was 93% adherent and the civamide cream 0.01% (control) group was 95% adherent. The civamide cream 0.075% group applied on average 2.4 g/day while the civamide cream 0.01% (control) group applied 2.6 g/day. They were also very adherent with their stable oral OA medication: the civamide cream 0.075% group was 95% adherent and the civamide cream 0.01% (control) group was 96% adherent.
Table 4 presents a summary of adverse events from the double-blind study that were determined by the investigators to be possibly or probably related to the study drug. Only those events deemed to be at least common in frequency (defined as occurring in > 1% of patients in either treatment group, in accord with MedDRA frequency convention) are listed. None of these events met the criteria for serious adverse events (SAE).
Summary of safety data for the double-blind study (12 weeks).
There were 229 civamide cream 0.075% patients (67%) and 181 civamide cream 0.01% (control) patients (52%) reporting at least 1 TEAE. The treatment groups differed significantly for the percentages of patients reporting a TEAE (p < 0.001) as compared by Fisher’s exact test due primarily to the higher number of application site reactions reported by the civamide cream 0.075% treatment group.
Application site burning sensations were the most frequently reported adverse events by both treatment groups, predominantly mild to moderate in severity. Burning sensations were initially reported on Day 1 by 18% and 4% of the civamide cream 0.075% and the civamide cream 0.01% groups, respectively. By Day 14 the corresponding numbers decreased to 10% and 3% and decreased further by Day 84 to 6% and 1% (Figure 6). Overall, 35% of the civamide cream 0.075% and 11% of the civamide cream 0.01% groups experienced burning sensations at least once during the study. The burning sensations were transient, with about 50% of the events lasting 0–30 minutes, about 23% of the events lasting 31–60 minutes, and the remaining 27% lasting > 1 hour.

Proportion (± SD) of safety population patients experiencing burning sensations during double-blind study treatment period.
A total of 15 patients reported 18 SAE. In the civamide cream 0.075% group, 7 patients (2%) reported 10 SAE. In the civamide cream 0.01% (control) group, 8 patients (2%) reported 8 SAE. There was no pattern to these events, none was reported by the investigator as related to the study drug, and none was the result of laboratory measurements, physical examination or vital signs.
Longterm study
A total of 305 of the patients (87%) used civamide cream 0.075% TID for 6 months and 284 (81%) applied civamide cream 0.075% for 12 continuous months. The safety population included all enrolled patients who received at least 1 dose of the study drug.
There were 247 patients (70%) who reported at least 1 TEAE. Events deemed to be at least common in frequency (> 1% of patients, in accord with MedDRA frequency convention) were application site burning in 76 patients (22%) and application site warmth in 14 patients (4%). None of these events met the criteria for SAE.
A total of 36 patients (10%) reported 41 SAE. There were 39 SAE deemed to be nonreportable (on the basis of Investigational New Drug Safety Reporting requirements in the US Code of Federal Regulations; 21 CFR 312.32). There was no pattern to these events, none was reported by the investigator as directly related to the study drug, and none was the result of laboratory measurements, physical examination or vital signs. Two SAE, while not considered to be study-drug related, were reportable. One was a 64-year-old man with a 40 pack-year history of smoking who was diagnosed with metastatic lung cancer during the study and was hospitalized prior to dying. He received civamide cream 0.075% in both studies. The other was an SAE of pancreatic cancer that occurred in a 72-year-old woman. She withdrew from the study and was treated with chemotherapy. She received civamide cream 0.075% in both studies.
DISCUSSION
The double-blind study was conducted in a population of patients already taking some form of systemic pharmacologic treatment for OA, but having inadequate response to that intervention. This represents a group for whom current therapeutic options are usually limited. Civamide cream 0.075% was shown to be effective in patients with greater pain levels, i.e., patients with a baseline WOMAC pain subscale score > 10. The demonstration of efficacy with civamide when used as adjunctive therapy is notable, as many clinical trials have shown that in this setting, responses to treatment are often less marked than when used as monotherapy, perhaps due to a ceiling effect on room for improvement in adjunctive use21. When used in a population already being treated, a greater treatment effect may be required than in a population not on a primary therapy for a positive outcome to be observed. Civamide at a strength of 0.01% was used to control for the transient burning sensation experienced with the test drug at the application site in some patients, and thus to better blind the study compared to the blinding of a true placebo or vehicle cream. This also made it more difficult to show a difference in efficacy between civamide cream 0.075% and control. As expected, the civamide cream 0.01% had activity in terms of pain relief as well (within-group reduction p < 0.001) over the 84 days of the study. However, even with this study design, the statistically significant and clinically relevant benefits of civamide cream 0.075% therapy were demonstrated with a significant difference in each of the 3 co-primary efficacy variables between baseline and Day 84 and with consistency at most other interim timepoints and secondary endpoints. Further, the robustness of the primary efficacy results was confirmed by the BOCF and WOCF sensitivity analyses.
It has been demonstrated as part of the investigation of the baseline-by-treatment interaction that civamide cream 0.075% shows better efficacy in those patients with higher baseline scores in both the WOMAC pain subscale and the WOMAC physical function subscale.
The clinical relevance and consistency of these results is further supported by the OMERACT-OARSI responder analysis results. The OMERACT-OARSI results are useful to clinicians because they demonstrate an overall clinical response, integrating individual outcome measurements. The OMERACT-OARSI supports the clinical significance of civamide cream 0.075% with a treatment difference between this treatment and control of 10% (p = 0.006). For patients with greater baseline pain severity, this treatment difference increases and is 27% (p < 0.001) for patients with severe pain (baseline pain > 13). This benefit of increased treatment effect in patients with greater symptoms is an additional clinically desirable benefit of civamide cream 0.075%.
Although designed primarily as a safety study, the longterm open-label study showed a maintenance of improvement in OA pain and SGE that was sustained throughout the year-long study, further corroborating the clinical benefits recorded in the double-blind study. The absence of positive findings for both the FCC and the SF-36v2 could be related to the 12-week duration of the double-blind study, which may have been too short to detect treatment differences.
The trial would have been improved if there had been collection of primary efficacy data at an earlier timepoint or time-points, prior to Day 21, in order to determine the onset of action of civamide cream 0.075%. It was also a design weakness in the year-long study that the efficacy scales used did not match those in the 12-week double-blind study. This would have permitted an easier comparison between the 2 studies. An aspect of clinical investigations with medications such as civamide cream 0.075% is that the burning sensation experienced by some subjects may potentially unblind and/or bias the subjects in their evaluations of clinical response. The 12-week trial was designed with a control arm of a low dose of civamide cream for this reason. Additionally, 2 analyses were conducted to evaluate whether burning sensation had an effect on clinical response: 1 analysis considered burning sensation as a factor in the efficacy results and another compared mean treatment differences for those who experienced burning sensation versus those who did not. The analyses demonstrated (1) no indications of any interactions of the civamide cream 0.075% treatment effect with the occurrence of burning sensation for any of the 3 co-primary efficacy measures in the study; and (2) that the civamide cream 0.075% treatment effect is similar whether or not the patient experienced a burning sensation during the course of the study.
Figures 3 and 5, displaying observed mean changes from baseline in WOMAC pain subscale scores and WOMAC physical function subscale scores, respectively, show only a small separation between active drug and vehicle. These graphs are not ideal, however, as they do not take into account the significant baseline-by-treatment interaction that occurred in the 12-week trial. To take the significant baseline-by-treatment interaction into account, one would have a unique graph for each baseline pain/physical function score, and the separation between active drug and vehicle would visibly increase from graph to graph as the baseline score increases. Figures 3 and 5 show the mean observed changes to depict the decrease in pain and physical function observed in our study, despite the limitation that they do not show the separation between active drug and vehicle that occurs at each baseline score.
The most commonly reported adverse event across all clinical studies conducted to date was a self-limited burning sensation at the application site that is most often mild to moderate in severity and transient, lasting seconds to minutes. In the double-blind trial, 18% of patients reported this burning sensation on the first day, with the daily frequency decreasing throughout the study to 10% at 14 days and 6% at 84 days of treatment. The burning sensation did not affect the tolerability of civamide cream 0.075%, as demonstrated by the low dropout rate in this double-blind study, i.e., 16% in the civamide cream 0.075% group and 8% in the control group, which compares favorably to other trials of this kind. Similarly, in the year-long open-label followup study, there was only a 19% dropout rate, again exceedingly low for any study of this duration.
The adverse event profile of civamide cream 0.075% is consistent with the fact that civamide is not systemically absorbed and has no direct internal organ effects. In this regard, civamide differs from acetaminophen, oral NSAID, and COX-2 inhibitors. Even topical NSAID have systemic absorption, albeit at lower levels than after oral administration, and still can be associated with organ-specific adverse effects22,23,24,25,26,27. Consequently, when compared to other agents used to treat OA pain, civamide cream has an overall excellent benefit/risk profile. This is also the case when considering opioid agents, used for the relief of severe arthritic pain, which can be associated with a high incidence of GI and CNS side effects24. Additionally, compared to the literature for topical capsaicin products28,29,30,31, civamide cream 0.075% has been shown to be accompanied by less burning sensation. Civamide cream 0.075% has clinical data for 1 year, whereas no other topical capsaicinoid does. Civamide cream 0.075% causes no measurable systemic absorption of civamide and therefore cannot lead to systemic toxicity.
In patients with OA who are experiencing significant pain despite oral medications, and especially patients with more severe symptoms, a clinician often considers additional medication. A topical treatment such as civamide cream 0.075% that has demonstrated efficacy without serious systemic safety concerns could be used adjunctively to oral therapies. In addition to providing incremental pain relief, civamide may also result in a reduction in the ongoing need for concomitant oral pain medication, thus reducing exposure to drugs (such as NSAID and COX-2) having dose-related unwanted effects.
Acknowledgment
The authors thank the investigators and their staffs who participated in the studies: C. Birbara, E.P. Boling, M.A. Borofsky, J.R. Caldwell, N. Chang, B.C. Corser, J.P. Donohue, H.F. Farmer, J.I. Fidelholtz, W.T. Garland, L.I. Gilderman, J.E. Greenwald, J.P. Hampsey, P.A. Holt, M. Hooper, R.M. Karr, R.A. Khairi, A.J. Kivitz, T.C. Klein, M.W. Layton, R.W. Levin, T.W. Littlejohn III, A.R. Mabaquiao, A. Mangione, H.W. Marker, B.A. McIntosh, D. Owens, B. Packman, S. Patel, D.S. Perling, B.C. Pogue, C.H. Pritchard, B.G. Rankin, S. Reddy, D.M. Rice, S. Roth, J. Rubino, L.A. Rudolph, M. Sack, D.R. Schumacher, D.J. Seiden, T.L. Shlotzhauer, S.L. Silverman, W.W. Storms, K.W. Strader, S. Touger, R.G. Trapp, D.R. Trotter, and B. Troupin. We also thank the patients who made these studies possible.
Footnotes
-
Supported by Winston Laboratories Inc. A.R. Sampson is a consultant to Winston Laboratories Inc.
- Accepted for publication September 20, 2011.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}