

Abstract
Objective. Immune complexes play an important role in the pathogenesis of primary Sjögren’s syndrome (pSS). Crosslinking of the neutrophil-specific Fc-γ receptor 3b (FCGR3B) facilitates immune complex clearance, and copy number variation (CNV) of the FCGR3B gene is known to reduce the uptake, and potentially clearance, of circulating immune complexes. Our objective was to determine whether FCGR3B CNV is a risk factor for pSS.
Methods. This was a cross-sectional study of patients with established pSS (n = 174) and population-matched controls (n = 162). FCGR3B CNV was determined by a quantitative real-time polymerase chain reaction assay, using genomic DNA as template and Taqman chemistry. Reactions were performed as a duplex, with RNAse P as the reference gene. Clinical and serological data were analyzed for their association with FCGR3B copy number (CN).
Results. Low FCGR3B CN (< 2 copies) was a risk factor for pSS in this cohort (p = 0.016), and combined results from this and a previous study yielded an overall OR of 2.3 (95% CI 1.3, 3.9, p = 0.003). Among patients with pSS in our cohort, low FCGR3B CN was not associated with anti-Ro ± La autoantibodies, but was associated with lower rheumatoid factor titers (p = 0.001) and serum IgG levels (p = 0.031).
Conclusion. We confirmed that, similarly to other systemic autoimmune diseases, FCGR3B CN is a genetic susceptibility factor for pSS. As in rheumatoid arthritis, the mechanism does not appear to be related to seropositivity for characteristic autoantibodies.
Primary Sjögren’s syndrome (pSS) is a common systemic autoimmune inflammatory condition. The predominant feature is failure of exocrine glands, although extraglandular manifestations, involving skin, lung, heart, kidneys, and nervous system, are common1,2. Autoantibodies targeting the ribonuclear proteins, Ro and La, are common in pSS and present in up to 80% of patient sera samples3. Although previously thought to be nonpathogenic “epiphenomena,” anti-Ro ± La are associated with systemic complications such as vasculitis, skin lesions, and neonatal heart block4,5. These autoantibodies are now thought to play a central role in the inflammatory response through immune complexes (IC) that are involved in cytokine release and complement activation6,7.
There is clearly a genetic component to susceptibility for pSS, or specifically, autoantibody-positive pSS8,9,10. Copy number variation (CNV) of the FCGR3B gene is a candidate gene of particular interest. CNV is a departure from the normal diploid number of genes (n = 2) that may arise from gene duplication and deletion events, and may contribute substantially to quantitative variation in gene expression. An increasing number of CNV have been characterized in the human genome, with implications for both evolution and disease susceptibility11. CNV has been identified in multiple genes within the Fc-γ receptor (FCGR) gene cluster on chromosome 1q2312. This cluster carries 5 highly homologous genes that encode for low affinity receptors for IgG-complexed antigens, which are expressed widely throughout the hematopoietic system. These low affinity FCGR are involved in the regulation of a multitude of innate and adaptive immune responses, with implications for both response to infection and susceptibility to autoimmunity13.
FCGR3B is an activating glycoprotein found only on the surface of human neutrophils that preferentially binds IgG14. Cross-linking between FCGR3B and IC initiates an effector response resulting in the clearance of IC15,16. CNV of FCGR3B has been well characterized, and there is a clear correlation between gene copy number and FCGR3B cell-surface expression, neutrophil adherence to IgG-coated surfaces, and uptake of IC17. Multiple studies have confirmed that low (< 2) FCGR3B copy number (CN) is a genetic susceptibility factor for systemic autoimmune diseases such as systemic lupus erythematosus (SLE)18,19,20 and rheumatoid arthritis (RA)21,22. Only 1 study has examined FCGR3B CN in pSS, reporting that, similar to their SLE cohort, both low (< 2) and high (> 2) FCGR3B CN was associated with pSS19.
We examined the association between FCGR3B CN and pSS in another cohort of patients.
MATERIALS AND METHODS
Study participants
The study population consisted of 174 white patients with pSS (90% females, median age 58 yrs, 84% seropositive for anti-Ro ± La autoantibodies), who met the revised 2002 American–European consensus research classification criteria for pSS23, and 162 white population-based controls (53% females, median age 56 yrs). Anti-Ro ± La autoantibody specificity and serum B cell activating factor (BAFF) levels were evaluated as described8,9,24, and patients were also assessed by case note review.
The study was conducted in accord with the Declaration of Helsinki and approved by the Central North Adelaide Health Service Ethics of Human Research Committee. All participants provided informed, written consent.
FCGR3B CNV determination
Genomic DNA was prepared by the salt precipitation method from fresh blood samples, as described10, and genomic FCGR3B CN was determined using a quantitative real-time polymerase chain reaction (qPCR) method21. Briefly, a duplex Taqman® CN assay was performed, using FCGR3B-specific primers (Applied Biosystems, Hs04211858, FAM-MGB dual labeled probe) and RNase P (Applied Biosystems, product 4403326, VIC-TAMRA dual labeled probe) as the reference assay. The assay was performed according to the manufacturer’s instructions and PCR were run on an Applied Biosystems 7300 Real Time PCR machine. All samples were tested in triplicate, and fluorescence signals were normalized to ROX dye. CN was determined using Copy Caller software (v.1.0, Applied Biosystems), and results were accepted only when calling confidence for discrete CN assignment was > 80%, and the ΔCq SD between replicates was < 0.20; otherwise, samples were retested.
The Taqman qPCR FCGR3B CN assay was initially validated against an endpoint PCR paralog ratio assay, measuring FCGR2C/FCGR2A CN ratios20, for which the signal intensity (peak height) for both amplicons (274 and 279 bp) was quantified through a multiplexed fluorescence capillary electrophoresis detection system (Qiaxcel). The rationale for this validation is that several studies have reported complete agreement (i.e., linkage disequilibrium) between FCGR2C and FCGR3B CN12,20. Three reference samples (1,2,3 CN) validated by this assay were included on each qPCR assay plate to control for possible batch effects.
Statistical analysis
The association between FCGR3B CN and disease susceptibility, and other dichotomous variables, was analyzed by chi-square and logistic regression analysis, with results reported as OR. Testing for ordinal relationships between FCGR3B CN and other variables was performed using the nonparametric gamma correlation coefficient, and proportional OR as appropriate. All analyses were performed using Statistica v6 (Statsoft).
A random effects metaanalysis of this study and a previous publication, examining the relationship between low CN of FCGR3B and pSS, was performed using the R metafor library25,26 and the restricted maximum likelihood method.
P values < 0.05 were considered to indicate statistical significance.
RESULTS
The number of genomic FCGR3B copies observed in our study varied from 0 to 4; however, 1−3 copies were the most common. One pSS patient carried no FCGR3B (null), and 1 patient and 1 control each carried 4 FCGR3B copies. Accordingly, the data were grouped for analysis into < 2, 2, and > 2 copies, respectively.
The distribution of FCGR3B CN variants was significantly different between pSS patients and controls (chi-square = 6.10, df = 2, p = 0.047; Table 1). Expressed as OR relative to the normal diploid 2 CN, low (< 2) CN was significantly increased in pSS (OR 2.6, 95% CI 1.2, 5.6, p = 0.016), whereas there was no difference in the frequencies of high (> 2) CN between pSS patients and controls (OR 1.1, 95% CI 0.5, 2.4, p = 0.80). Although the sample sizes in this study were comparatively small, a retrospective power analysis indicated that there was sufficient power (79.6% 1-sided test, 69.5% 2-sided test) to detect a difference in low FCGR3B CN frequency between patients and controls.
Low FCGR3B genomic copy numbers (< 2 CN) is associated with primary Sjögren’s syndrome (pSS), but not specifically Ro + La autoantibody (seropositive) pSS. The distribution fo FCGR3B CN was significantly different between pSS patients and controls (p = 0.047). When expressed as OR relative to the normal diploid 2 CN, low (< 2) CN was significantly increased in pSS (p = 0.016), wheras there was no difference in the frequencies of high (> 2) CN between pSS patients and controls (p = 0.80). The frequency distribution was also different twhen comparing seropositive versus seronegative pSS patients (p = 0.042), which was primarily attributable to a higher proportion of high (> 2) CN in seronegative patients with pSS. Thee was, however, no suggestion that low (< 2) CN is preferentially associated with Ro + La seropositive pSS.
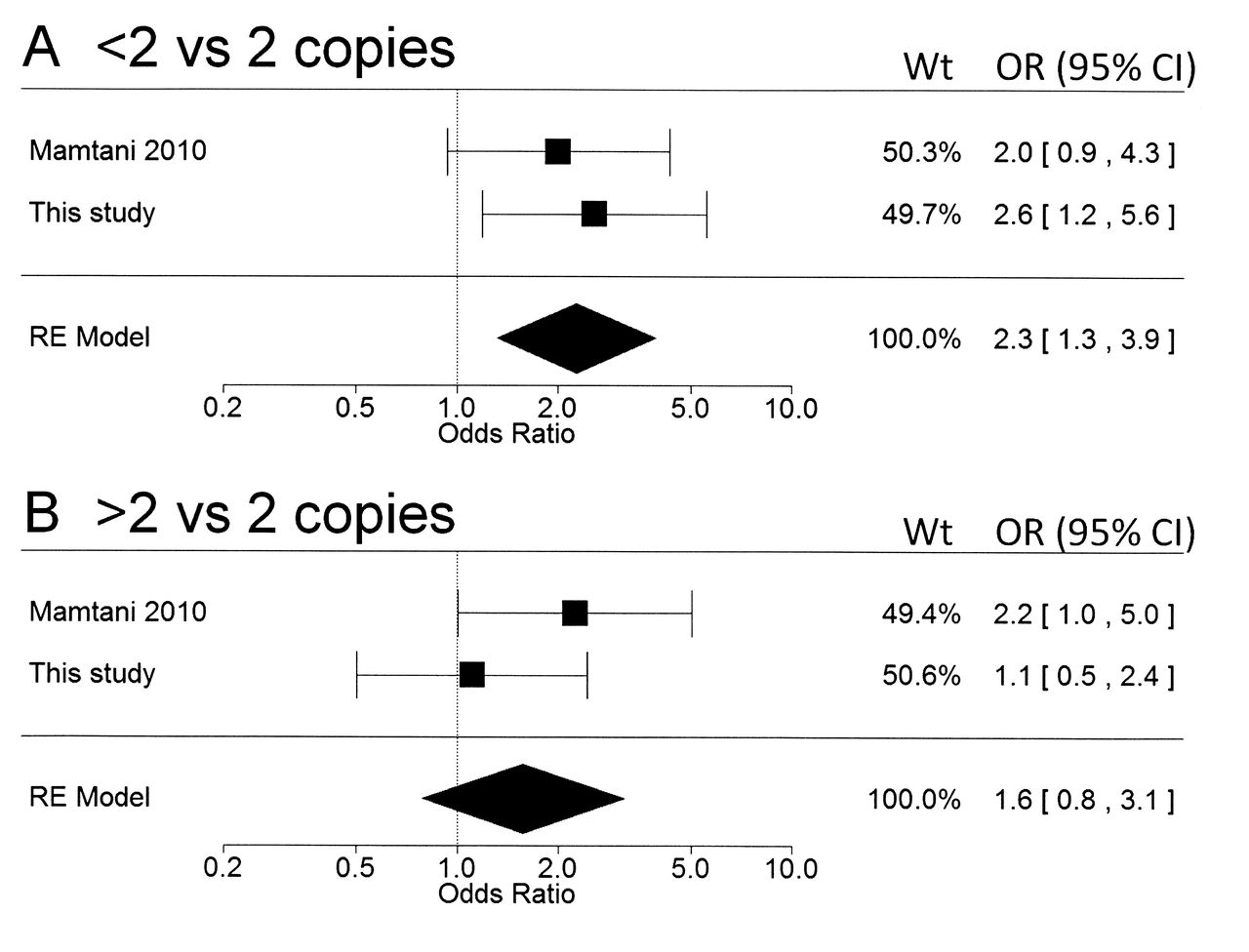
One previous study evaluated FCGR3B CN in pSS19. In a metaanalytic approach, the combined results from both studies (Figure 1) yielded OR of 2.3 (95% CI 1.3, 3.9, p = 0.003) for low (< 2) CN, and 1.6 (95% CI 0.8, 3.1, p = 0.20) for high (> 2) CN, both expressed relative to the normal diploid 2 CN. Therefore, the association between low FCGR3B CN is confirmed in 2 separate studies, but the additional putative association with high FCGR3B CN19 remains unconfirmed. Our observation that high (> 2) FCGR3B CN was more frequent in patients with seronegative pSS (Table 1) may be relevant in this context, although the number of seronegative patients was very small.

Random effects (RE) metaanalysis of the association between FCGR3B copy number variations and primary Sjögren’s syndrome. The 2 studies available were Mamtani 201019 and the current study. (A) < 2 versus 2 FCGR3B copies; p = 0.003. (B) > 2 versus 2 FCGR3B copies; p = 0.20. RE model indicates the OR (95% CI), estimated by restricted maximum likelihood, for the 2 studies combined.
Given that FCGR play an important role in the clearance of IC, it is relevant to consider whether low FCGR3B CN, as a risk factor for pSS, is specifically associated with seropositivity for Ro ± La autoantibodies within patients with pSS. The distribution of FCGR3B CN in Ro ± La-seronegative versus seropositive patients with pSS is also shown in Table 1. Although the number of patients with seronegative pSS in this study was limited, the frequency distribution was different between the 2 groups (p = 0.042), which was primarily attributable to a higher proportion of high (> 2) CN in patients with seronegative pSS. The relevance of this observation is uncertain, given the low numbers of patients with seronegative pSS. There was, however, no suggestion that low FCGR3B CN is preferentially associated with Ro ± La-seropositive pSS.
In terms of other clinical/serological features, low FCGR3B CN was associated with lower rheumatoid factor (RF) titers and lower serum IgG levels (Table 2, Figure 2). There was also a trend for an inverse ordinal relationship between FCGR3B CN and the proportion of patients with a history of persistent, presumed autoimmune neutropenia, although the number of such patients was low (26/254 = 17%) and this analysis did not achieve statistical significance (p = 0.12; Table 2, Figure 2). There was no observed relationship with age of onset, sex, Schirmer’s test results, sialometry results, or C4 or serum BAFF levels (data not shown).

Box plots of FCGR3B copy number (CN) by (A) rheumatoid factor titer (n = 140) and (B) serum IgG (n = 138). The width of the box plots reflects the number of observations. It is evident that the ordinal trends reported in Table 3 are primarily due to low (< 2) FCGR3B CN, which is associated with lower RF titers and lower serum IgG. (C) The prevalence of low FCGR3B CN is slightly higher in patients with a history of neutropenia, and the prevalence of high FCGR3B CN slightly lower, indicative of an inverse ordinal trend.
Within patients with primary Sjögren’s syndrome there is a positive ordinal relationship between FCGR3B copy number and rheumatoid factor (RF) titer and serum IgG levels, and an inverse ordinal trend with the risk of neutropenia.
DISCUSSION
We have confirmed that low FCGR3B CN is a genetic susceptibility factor for pSS. Our results for the association with low FCGR3B CN were very similar to the single previous study of patients with pSS19, and collectively, the OR for the association with pSS (relative to the normal diploid 2 CN) was estimated as 2.6 (95% CI 1.2, 5.6, p = 0.003). Therefore low FCGR3B CN may be a common genetic risk factor for systemic autoimmune diseases such as SLE18,19,20, RA21,22, and pSS. However, it is also clear that a relatively low proportion (< 20%) of patients with systemic autoimmune diseases appear to carry this genetic risk factor. This may be indicative of underlying disease heterogeneity, in which low FCGR3B CN is a risk factor in an as-yet unidentified subgroup of patients.
The exposure of autoantigens in pSS is thought to occur during apoptosis of salivary gland acinar and ductal epithelial cells, which is accompanied by translocation of autoantigens to apoptotic blebs. This local event may initiate an autoimmune reaction that becomes systemic through upregulation of type I interferon signature and abnormal expression of BAFF7, and lymphocytic invasion and/or IC-mediated inflammation in other organ systems may result. Circulating IC can be demonstrated in a majority of patients with pSS, and anti-Ro-containing IC are related to continuing systemic immune activation27,28,29. As FCGR form an important pathway for IC clearance15,16,17, it was therefore surprising that we were unable to demonstrate a relation between low FCGR3B CN and anti Ro ± La autoantibodies in pSS. In fact we observed that low FCGR3B CN was associated with lower RF titers and serum IgG levels in patients with pSS. We previously demonstrated that diversification of the Ro ± La autoantibody response in patients with pSS is under genetic control, and correlates strongly with RF titers and serum IgG levels5,10. While our findings in relation to low FCGR3B CN are only exploratory, and require replication in future studies, collectively, these results suggest that the autoimmune response may be somewhat attenuated in patients who carried this risk factor. In keeping with our findings, no study to date has been able to demonstrate a relationship between low FCGR3B CN and disease-specific autoantibodies in systemic autoimmunity. Therefore, the evidence to date suggests that the pathogenetic mechanisms underpinning the relationship between low FCGR3B CN and systemic autoimmunity do not specifically relate to clearance of autoantigen-containing IC. One possibility is that, as FCGR3B is an important defense mechanism for the clearance of microorganisms, low FCGR3B CN may delay the resolution of infections, such as Epstein-Barr virus (EBV), which may act as a trigger for systemic autoimmunity in some patients30. In this context, it is perhaps relevant that the single FCGR3B-null, seronegative patient with pSS in this study had a history of recurrent EBV infections, and our observation that low FCGR3B CN is associated with lower IgG levels is also consistent with this hypothesis.
Other pathogenetic mechanisms may also be relevant, but unfortunately the role of neutrophils and FCGR3B in pSS has not been studied extensively. The expression of FCGR3B is upregulated in the saliva of patients with pSS, suggestive of neutrophil activation in salivary glands31. Neutropenia may be a relevant hematological finding in pSS32, and our data were suggestive, but not conclusive, that low FCGR3B CN may be a risk factor for neutropenia in patients with pSS. Clinically relevant autoantibodies targeting the human neutrophil FCGR have been described in pSS33,34, and soluble FCGR3B may act as a ligand for complement receptor activation35. However, factors that influence the relative expression of sFCGR3B versus membrane-bound FCGR3B, in both health and disease, are not understood.
It is also not clear whether the association with systemic autoimmunity can be specifically, or solely, attributable to FCGR3B CN. The FCGR gene cluster is a complex genomic region, carrying multiple genes, and is characterized by both single-nucleotide polymorphism (SNP) and CNV polymorphism and linkage disequilibrium. For example, FCGR3B and FCGR2C CNV appear to be in complete linkage disequilibrium12,20, and FCGR2C, although a pseudogene in some individuals, may be expressed in a wider range of cell types, including dendritic and natural killer (NK) cells12. Recent evidence also suggests that the FCGR2C-FCGR3B insertion/deletion extends into FCGR2B, the only inhibitory FCGR, and probably deletes a negative regulatory element in the FCGR2B promoter in NK cells36. Studies have also implicated SNP in multiple FCGR genes with susceptibility to systemic autoimmunity16, and 1 study has demonstrated that both FCGR3B CNV and SNP polymorphism contribute to SLE susceptibility37. The future challenge will be to integrate CNV and SNP data into FCGR gene-cluster haplotypes to systematically evaluate and contrast disease associations.
Our results confirm that, similar to other systemic autoimmune diseases, low FCGR3B CN is a genetic risk factor for pSS. However, the underlying pathogenetic mechanism does not appear to relate specifically to clearance of immune complexes formed by anti-Ro ± La autoantibodies. Further studies on immune complex clearance in systemic autoimmunity, with specific respect to the role of neutrophils, are warranted, in addition to careful clinical characterization of patients who carry this genetic risk.
Acknowledgment
The authors thank Angela Berry for technical assistance.
Footnotes
-
Supported by grants from the Norwegian Rheumatism Association, the North Norwegian Health Authority, and the Hospital Research Foundation.
- Accepted for publication July 6, 2012.








{kind=link}
{kind=link}