
Abstract
Objective. The aim of our study was to investigate the effect of an adenosine A2A receptor agonist, 2-[p-(2-carboxyethyl)phenylethylamino]-50 ethylcarboxamidoadenosine (CGS 21680), on modulation of the inflammatory response in mice subjected to collagen-induced arthritis (CIA).
Methods. CIA was induced by intradermal injection of 100 μl of emulsion containing 100 μg of bovine type II collagen (CII) and complete Freund’s adjuvant (CFA) at the base of the tail. On Day 21, a second injection of CII in CFA was administered. Immunized mice developed erosive hind paw arthritis. Macroscopic clinical evidence of CIA first appeared as periarticular erythema and edema in the hind paws. The incidence of CIA was 100% by Day 27 in the CII challenged mice and the severity of CIA progressed over a 35-day period, with radiographic evaluation revealing focal resorption of bone. The histopathology of CIA included erosion of cartilage at the joint margins.
Results. Treatment of mice with CGS 21680 starting at the onset of arthritis (Day 25) ameliorated the clinical signs at Days 26–35 and improved histological status in the joint and paw. The degree of oxidative and nitrosative damage was significantly reduced in CGS 21680-treated mice as indicated by elevated levels of malondialdehyde, formation of nitrotyrosine, and activation of poly(ADP-ribose) polymerase. Plasma levels of proinflammatory cytokines such as tumor necrosis factor, interleukin 1ß (IL-1ß) and IL-6 were also reduced by CGS 21680. Treatment with CGS 21680 also decreased the expression of inducible nitric oxide synthase and cyclooxygenase-2.
Conclusion. We demonstrate that CGS 21680 exerts an antiinflammatory effect during chronic inflammation and ameliorates the tissue damage associated with CIA.
- ARTHRITIS
- CGS 21680
- INFLAMMATION
- APOPTOSIS
- ADENOSINE
Rheumatoid arthritis (RA) is an autoimmune disease characterized by the sequestration of various leukocyte subpopulations within both the developing pannus and the synovial space. The chronic expression of this disease results in multiple joint inflammation with subsequent destruction of joint cartilage and erosion of bone. While this disease has a world-wide distribution, its pathogenesis is not clearly understood1. Type II collagen-induced arthritis (CIA) in the mouse has proven to be a useful model of RA, as it possesses many of the cell and humoral immunity characteristics found in human RA2. The pathogenesis of CIA is dependent upon the host’s response to type II collagen challenge and subsequent generation of antibodies that recognize collagen-rich joint tissue2. The chronic activities initiated by immune complexes trigger a variety of cell-mediated and humoral events. Moreover, the recruitment and activation of neutrophils, macrophages, and lymphocytes into joint tissues and the formation of pannus are hallmarks of the pathogenesis of both CIA and human RA. While the contribution of inflammatory leukocytes to the progression of experimental arthritis and human RA is unquestioned, the mechanisms whereby these leukocytes are recruited to the inflamed joint are still not fully known.
Recently, it has been demonstrated that interleukin 8 (IL-8), macrophage inflammatory proteins (MIP-lα), MIP-1ß, and regulated upon activation, normal T cell expressed and secreted (RANTES) are differentially chemotactic for lymphocyte subsets3. Chemokines may play a prominent role in RA, as neutrophil and mononuclear cell stimulation and activation are prevalent in this disease.
Evidence has suggested the involvement of adenosine receptors in the process of inflammation4,5. Adenosine exerts its cellular activity through one of 4 G-protein-coupled receptors: A1, A2A, A2B, and A3. The antiinflammatory effects of adenosine are generally attributed to occupancy of A2A receptors6. The stimulation of A2A receptors limits macrophage proinflammatory cytokine production7, reduces adhesion molecule expression on endothelial cells8, and suppresses generation of superoxide anion and leukotriene synthesis by neutrophils9. Studies have tested the potential of A2A receptor activation to prevent local damage following ischemia/reperfusion insults. These studies found that A2A receptor activation protects the lung10, liver11, kidney12, and spinal cord13 following locally induced ischemia/reperfusion injury.
Varani and colleagues demonstrated that the upregulation of A2A and A3 receptor in patients with early RA and in patients with RA treated with methotrexate was associated with high levels of tumor necrosis factor (TNF-α) and nuclear factor-κB (NF-κB) activation4. Varani and colleagues also pointed out that treatment with anti-TNF-α normalized A2A and A3 receptor expression and function4.
The objective of our study was to investigate whether 2-[p-(2 carboxyethyl)phenylethylamino]-50 ethylcarboxamidoadenosine (CGS 21680) ameliorated the development of arthritis caused by injection of collagen type II (CII) in mice. CGS 21680 is considered an A2A adenosine receptor agonist but there is some uncertainty about its target. Indeed, in humans CGS 21680 is highly specific for A2A versus A2B, but it shows only a 2–3-fold specificity versus A3 and only 10-fold versus A114; and there is a report of an action of CGS 21680 on A1 receptor in mice in which this drug exhibits binding characteristics that are not compatible with adenosine A2A receptor binding15. Moreover, the affinity of the drug for the different murine adenosine receptors remains unknown and might differ from what has been determined for the human receptors. The affinity of different adenosine agonists is shown to be sometimes very different between human and rat receptors, demonstrating that there is difference between species for the binding of these drugs14.
We evaluated the following endpoints of the inflammatory process: (1) clinical score; (2) body weight; (3) expression of inducible nitric oxide synthase (iNOS), cyclooxygenase 2 (COX-2), and pJNK; (4) nitrotyrosine formation and activation of the nuclear enzyme poly(ADP-ribose) polymerase (PARP); (5) lipid peroxidation; (6) cytokine and chemokine production; (7) neutrophil infiltration; (8) bone erosion (on radiography); and (9) joint histopathology.
MATERIALS AND METHODS
Animals
DBA/1J mice (9 weeks of age; Harlan-Nossan, Milan, Italy) were used for these studies. Animals were housed in a controlled environment and provided with standard rodent chow and water. Animal care was in compliance with Italian regulations on protection of animals used for experimental and other scientific purposes (D.M. 116192) as well as with the EEC regulations (O.J. of E.C. L358/1 12/18/1986).
Experimental groups
Mice were divided into the following 4 experimental groups.
CIA-Control: mice were subjected to CIA and administered 200 μl of 10% DMSO solution intraperitoneally (vehicle for CGS 21680) every 24 h, starting from Day 25 to Day 35 (n = 20).
CIA-CGS 21680; mice subjected to CIA were administered CGS 21680 0.1 mg/kg intraperitoneally every 24 h, starting from Day 25 to Day 35 (n = 20). Sham-Control: mice subjected to an intradermal injection at the base of the tail of 100 μl of 0.01 M acetic acid instead of the emulsion containing 100 μg of CII were treated with 200 μl of 10% DMSO solution intraperitoneally (vehicle for CGS 21680) every 24 h starting from Day 25 to Day 35 (n = 20). Sham-CGS 21680: mice subjected to an intradermal injection at the base of the tail of 100 μl of 0.01 M acetic acid instead of the emulsion containing 100 μg of CII were administered CGS 21680 0.1 mg/kg intraperitoneally every 24 h starting from Day 25 to Day 35 (n = 20).
The dose of CGS 21680 used to reduce joint injury was based on findings of a previous study16.
Induction of collagen-induced arthritis
Bovine CII was dissolved in 0.01 M acetic acid at a concentration of 2 mg/ml by stirring overnight at 4°C. Dissolved CII was frozen at −70°C until use. Complete Freund’s adjuvant (CFA) was prepared by addition of Mycobacterium tuberculosis H37Ra at a concentration of 2 mg/ml. Before injection, CII was emulsified with an equal volume of CFA. CIA was induced as described17. On Day 1, mice were injected intradermally at the base of the tail with 100 μl of the emulsion containing 100 μg CII. On Day 21, a second injection of CII in CFA was administered.
In addition, on Day 1 and on Day 21 the sham-control and sham-CGS 21680 groups also received, instead of collagen, 100 μl of 0.01 M acetic acid.
Clinical assessment of CIA
Development of arthritis in mice from all experimental groups was evaluated daily starting from Day 20 after the first intradermal injection by using a macroscopic scoring system: 0 = no signs of arthritis; 1 = swelling and/or redness of the paw or one digit; 2 = 2 joints involved; 3 = more than 2 joints involved; and 4 = severe arthritis of the entire paw and digits. Arthritic index for each mouse was calculated by summing the 4 scores of individual paws. Clinical severity was also determined by quantifying the change in the paw volume using plethysmometry (model 7140; Ugo Basile, Comeria, Italy)17.
Histological examination
On Day 35, the animals were sacrificed under anesthesia (sodium pentobarbital 45 mg/kg, intraperitoneally), and paws and knees were removed and fixed in 10% formalin. The paws were then trimmed, placed in decalcifying solution for 24 h, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin/eosin and Masson’s trichrome stain, then studied using light microscopy (Dialux 22, Leitz). Arthritis damage (histological damage score) was evaluated and scored by an investigator blinded to the treatment regime. The following morphological criteria were considered: score 0, no damage; score 1, edema; score 2, presence of inflammatory cells; score 3, bone resorption.
Radiography
The mice were anesthetized with sodium pentobarbital (45 mg/kg, intraperitoneally). Mice were placed on a radiographic box 90 cm from the x-ray source. Radiographic analysis of normal and arthritic rat hind paws was performed (Philips X12; Germany) with a 40 kW exposure for 0.01 s. An investigator blinded to the treatment regime performed radiograph scoring. The following radiographic criteria were considered: score 0, no bone damage; score 1, tissue swelling and edema; score 2, joint erosion; score 3, bone erosion and osteophyte formation.
Immunohistochemical localization of nitrotyrosine, PARP, iNOS, COX-2, and pJNK
On Day 35, the joints were trimmed and placed in decalcifying solution for 24 h and 8-μm sections were prepared from paraffin-embedded tissues. After deparaffinization, endogenous peroxidase was quenched with 0.3% H2O2 in 60% methanol for 30 min. The sections were permeabilized with 0.1% Triton X-100 in phosphate buffered saline (PBS) for 20 min. Nonspecific adsorption was minimized by incubating the section in 2% normal goat serum in PBS for 20 min. Endogenous biotin or avidin binding sites were blocked by sequential incubation for 15 min with avidin and biotin. Sections were incubated overnight with (1) anti-rabbit polyclonal antibody directed at iNOS (1:1000 in PBS, vol/vol; DBA, Milan, Italy), or (2) anti-COX-2 goat polyclonal antibody (1:500 in PBS, vol/vol), or (3) anti-nitrotyrosine rabbit polyclonal antibody (1:1000 in PBS, vol/vol), or (4) anti-poly(ADP-ribose) goat polyclonal antibody rat (1:500 in PBS, vol/vol), or (5) with anti-pJNK mouse polyclonal antibody (1:500 in PBS, vol/vol). Controls included buffer alone or nonspecific purified rabbit IgG. Specific labeling was detected with biotinylated pan-specific antibody (horse anti-mouse/rabbit/goat IgG; Vectastain ABC, DBA, Milan, Italy).
The counterstain was developed with DAB (brown color) and nuclear fast red (red background). In order to confirm that the immunoreaction for the nitrotyrosine was specific, some sections were also incubated with the primary antibody (anti-nitrotyrosine) in the presence of excess nitrotyrosine (10 mM) to verify the binding specificity. To verify the binding specificity for PARP, pJNK, COX-2, and iNOS, some sections were also incubated with only the primary antibody (no secondary) or with only the secondary antibody (no primary). In these situations, no positive staining was found, indicating that the immunoreaction was positive in all the experiments. Immunocytochemistry photographs (N = 5) were assessed by densitometry using an imaging densitometer (AxioVision, Zeiss, Milan, Italy) with computer software.
Western blot analysis for iNOS and COX-2
iNOS and COX-2 expression was detected as described18 in joint extracts by Western blot analysis. At specified times, joint tissues from left hind paws were collected and frozen until used. The frozen joint tissues were pulverized, and protein was extracted using RIPA buffer [50 mmol/l Tris-HCl, pH 7.5, 150 mmol/l NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS)]. Protein concentration was estimated with the DC Protein Assay kit (Bio-Rad, Milan, Italy). Equal amounts of protein samples (80 μg/lane) from pooled joint extracts were fractionated by 8% SDS-polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane in transfer buffer (39 mmol/l glycine, 48 mmol/l Tris base, 0.037% SDS, 20% methanol) at 100 mA for 2 h. The membranes were blocked in Tris-buffered saline containing 0.1% Tween 20 and 5% dry milk for 1 h at room temperature and subsequently probed overnight at 4°C with mouse monoclonal anti-iNOS (1:10,000) or anti-COX-2 (1:500) antibodies (in PBS, 5% wt/vol nonfat milk and 0.1% Tween-20). Anti-α-tubulin monoclonal antibody (Calbiochem, San Diego, CA, USA) was used as control. Immunoreactive protein bands were visualized using horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000) followed by enhanced chemiluminescence. The protein bands of iNOS and COX-2 on X-Omat films were quantified by scanning densitometry (Imaging Densitometer GS-700; Bio-Rad, Hercules, CA, USA).
Measurement of cytokines
TNF-α, IL-6, and IL-1ß levels were evaluated in the plasma from CIA mice as described19. Briefly, the assay was carried out using a colorimetric commercial ELISA kit (Calbiochem-Novabiochem Corp., Milan, Italy) with a lower detection limit of 10 pg/ml.
Measurement of chemokines
Levels of murine chemokine MIP-1α and MIP-2 were measured in the aqueous joint extracts. Briefly, joint tissues were prepared by first removing the skin and separating the limb below the ankle joint. Joint tissues were homogenized on ice in 3 ml lysis buffer (PBS containing 2 mM PMSF, and 0.1 mg/ml each (final concentration) of aprotinin, antipain, leupeptin, and pepstatin A using a Polytron (Brinkmann Instruments, Westbury, NY, USA). The homogenized tissues were then centrifuged at 2000 g for 10 min. Supernatant was sterilized with a millipore filter (0.2 μm) and stored at −80°C until analyzed. The extracts usually contained 0.2–1.5 mg protein/ml, as measured by protein assay kit (Pierce Chemical Co., Rockford, IL, USA). The levels of MIP-1α and MIP-2 were quantified using a modification of a double-ligand method20. Briefly, flat-bottom 96-well microtiter plates were coated with 50 μl/well rabbit anti-cytokine antibodies (1 μg/ml in 0.6 mol/l NaCl, 0.26 mol/l H3PO4, and 0.08 N NaOH, pH 9.6) for 16 h at 4°C, and then washed with PBS, pH 7.5, 0.05% Tween 20 (wash buffer). Nonspecific binding sites on microtiter plates were blocked with 2% bovine serum albumin in PBS and incubated 90 min at 37°C. Plates were rinsed 4 times with wash buffer, and diluted aqueous joint samples (50 μl) were added, followed by incubation for 1 h at 37°C. After washing of plates, chromogen substrate was added. The plates were incubated at room temperature to the desired extinction, then the reaction was terminated with 50 μl/well of 3 M H2SO4 solution. The plates were read at 490 nm in an ELISA reader. This ELISA method consistently had a sensitivity limit of ∼30 pg/ml.
Myeloperoxidase (MPO) assay
Neutrophil infiltration to the inflamed joints was indirectly quantified using an MPO assay, as described for neutrophil elicitation21.
Malondialdehyde (MDA) measurement
Plasma MDA levels were determined as an indicator of lipid peroxidation22. An aliquot (100 μl) of plasma collected at the specified time was added to a reaction mixture containing 200 μl of 8.1% SDS, 1500 μl of 20% acetic acid (pH 3.5), 1500 μl of 0.8% thiobarbituric acid, and 700 μl distilled water. Samples were then heated for 1 h at 95°C and centrifuged at 3000 g for 10 min. The absorbance of the supernatant was measured by spectrophotometer at 650 nm.
Materials
CGS 21680 was obtained from Merck Biosciences (Calbiochem, Beecham, Nottingham, UK). Unless otherwise stated, other compounds were obtained from Sigma-Aldrich Company (Milan, Italy). All chemicals were of the highest commercial grade available. All stock solutions were prepared in nonpyrogenic saline (0.9% NaCl; Baxter Healthcare Ltd., Thetford, Norfolk, UK) or 10% DMSO (Sigma-Aldrich).
Data analysis
All values in the figures and text are expressed as mean ± standard error (SEM) of n observations. For the in vivo studies, n represents the number of animals studied. In the experiments involving histology or immunohistochemistry, the figures shown are representative of at least 3 experiments (histological or immunohistochemistry coloration) performed on different experiment days on the tissue sections collected from all the animals in each group. Data sets were examined by one- or 2-way analysis of variance, and individual group means were then compared with Student’s unpaired t test. For the arthritis studies, Mann-Whitney U test (2-tailed, independent) was used to compare medians of the arthritic indices23. A p value < 0.05 was considered significant.
RESULTS
Effect of CGS 21680 on joint injury during experimental arthritis
CIA developed rapidly in mice immunized with CII and clinical signs of disease (periarticular erythema and edema; Figure 1B) first appeared in hind paws between 24 and 26 days post-challenge (Figure 1D), leading to 100% incidence of CIA at Day 28 (Figure 1D). Hind paw erythema and swelling increased in frequency and severity in a time-dependent manner with maximum arthritis indices of approximately 10 observed between Day 29 and 35 post-immunization (Figure 1D) in CIA-control mice. CGS 21680 treatment demonstrated a significant reduction of joint inflammation, as identified by a significant reduction in the incidence of arthritis (Figure 1C). CIA-CGS 21680 mice showed a 40% reduction in the development of arthritis and a significantly lower arthritis index compared to CIA-control mice (Figure 1D), suggesting a possible adenosine receptor desensitization. There was no macroscopic evidence of either hind paw erythema or edema in the sham-control group (Figure 1A, 1D) or the sham-CGS 21680 group (data not shown).
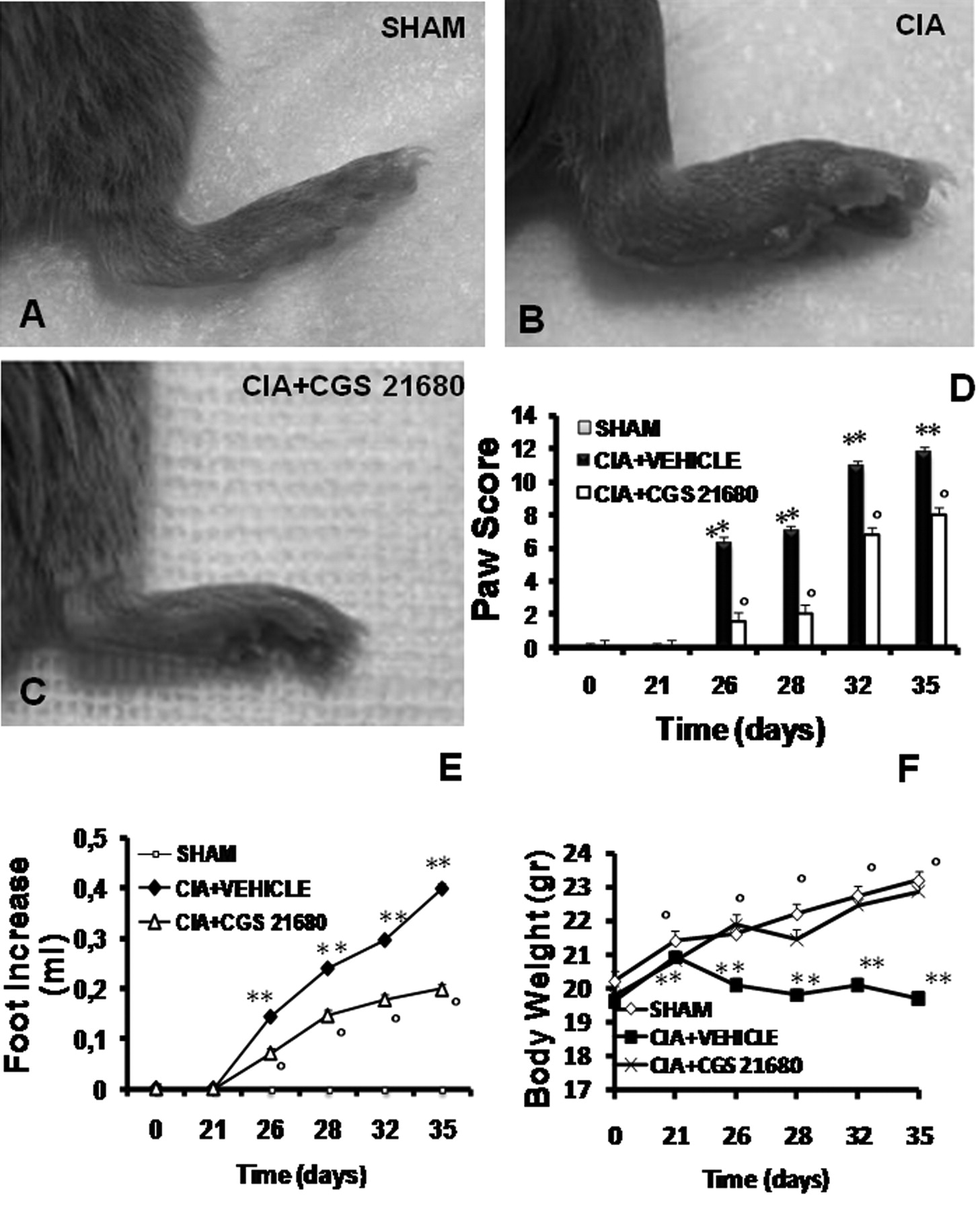
Effect of CGS 21680 treatment on the clinical expression of CIA, on the secondary lesion, and on body weight gain in CIA. A: No clinical signs were observed in sham-mice. B: CIA developed rapidly in mice immunized with CII, with clinical signs such as periarticular erythema and edema. D: Hind paw erythema and swelling increased in frequency and severity in a time-dependent mode. C: CIA-CGS 21680 mice had a significant reduction in the clinical signs of CIA. E: Swelling of hind paws over time was measured at 2-day intervals. F: Beginning on Day 25, the CII-challenged mice gained significantly less weight than the sham-control mice and this trend continued through Day 35. CIA-CGS 21680 mice demonstrated significantly reduced weight loss (F) and less paw edema (E). Data shown here are representative of all the animals in each group. Values are means ± SEM of 20 animals for each group. **p < 0.01 vs sham-control. °p < 0.01 vs CIA.
Figure 1E demonstrates a time-dependent increase in hind paw volume (each value represents the mean of both hind paws). The CIA-CGS 21680 mice showed a significant reduction of paw edema formation compared to CIA-control mice (Figure 1E). No increase in hind paw volume over time was observed in the sham-control mice (Figure 1E) or in sham-CGS 21680 mice (data not shown).
The rate and the absolute gain in body weight were comparable in sham-control and CIA-control mice in the first week (Figure 1F). From Day 25, the CII-challenged mice gained significantly less weight than the sham-control mice, and this trend continued through to Day 35. CGS 21680 treatment determined a significant increase of the weight gain compared with the vehicle treatment in CIA-control mice (Figure 1F).
Histological evaluation (on Day 35) of joints from CIA-control mice (Figure 2B, 2G) revealed signs of severe arthritis, with inflammatory cell infiltration and bone erosion. The histological alterations of joint were significantly reduced in CGS 21680-treated mice (Figure 2C, 2G). Moreover, Masson trichrome stain revealed decreased collagen (blue stain) in bone and cartilage of arthritic joints due to bone erosion and cartilage degradation in CIA-control mice (Figure 2E, 2G). The alterations of joints were significantly reduced in CGS 21680-treated mice (Figure 2F, 2G). A radiographic examination of knee joint and femoral growth plate in the femur from vehicle-treated mice at 35 days post-CII immunization revealed bone erosion (Figure 3B, 3D). Significantly less bone resorption was observed in the CGS 21680-treated mice (Figure 3C, 3D). There was no evidence of pathology in sham-control mice (Figures 2A, 2D, 2G; 3A, 3D) and in sham-CGS 21680 mice (data not shown).
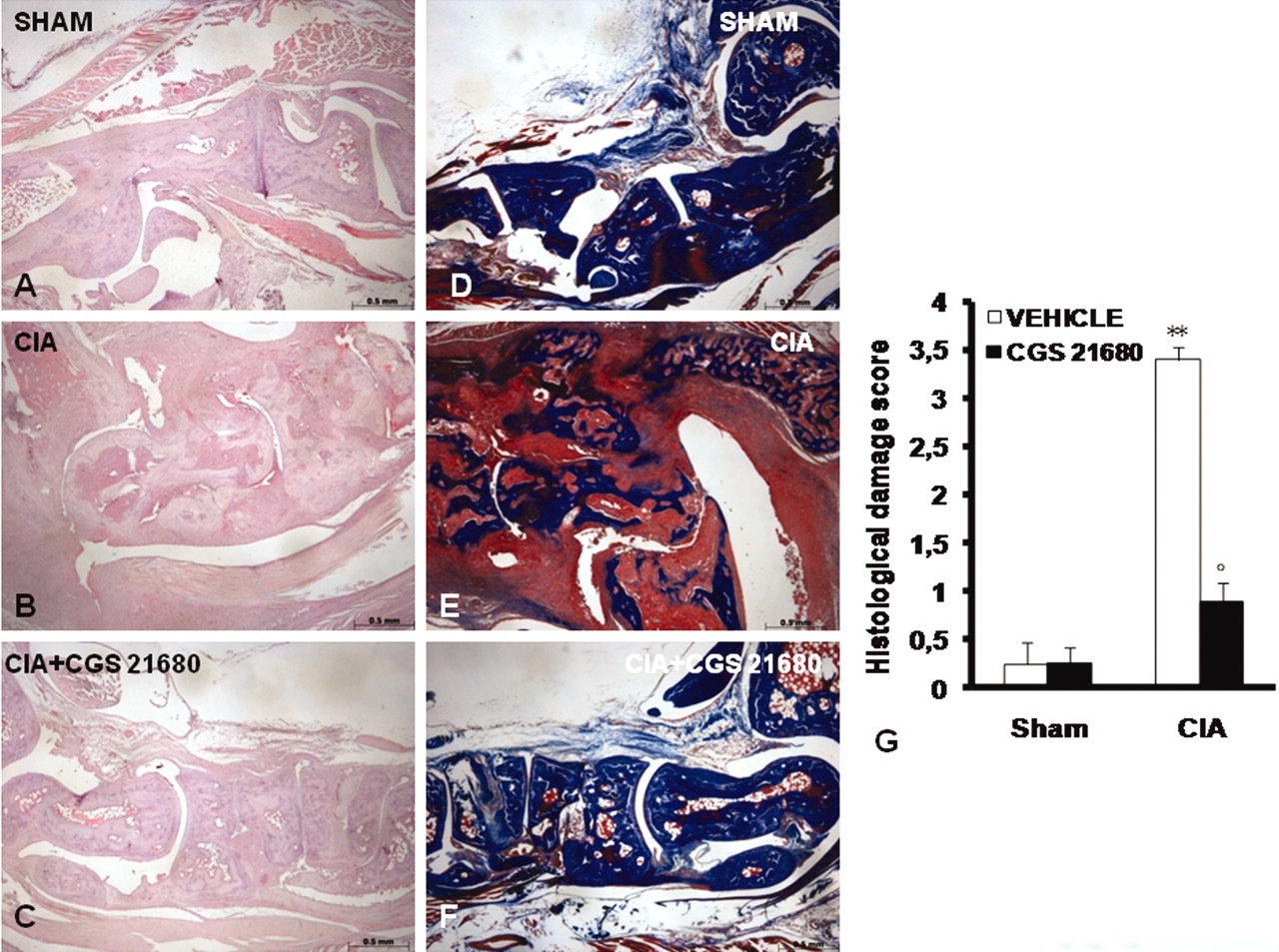
Morphologic changes of CIA. Representative hematoxylin/eosin-stained joint section was examined by light microscopy. Histological evaluation of joint (on Day 35) from CIA-control mice (B, G) revealed signs of severe arthritis, with inflammatory cell infiltration and bone erosion. Histological joint alterations were significantly reduced in tissues from CIA-CGS 21680 mice (C, G). Masson’s trichrome stain reveals decreased collagen (blue stain) in bone and cartilage of arthritic joint due to bone erosion and cartilage degradation in CIA-control mice (E, G). The joint alterations were significantly reduced in CGS 21680-treated mice (F, G). There was no evidence of pathology in the sham-control mice (A, D, G). Figure is representative of at least 3 experiments performed on different days. Values are means ± SEM of 20 animals for each group. **p < 0.01 vs sham-control. °p < 0.01 vs CIA.

Radiographic progression of CIA in the tibiotarsal joint of mice with CIA. There is no evidence of pathology in the femoral growth plate of normal mice (A, D) or in the tibiotarsal joints (A, D). Hind paws from CII-immunized (35 days) vehicle-treated mice showed bone resorption in the femoral growth plate of normal mice (B, arrow; D) as well as in the tibiotarsal joints (B, arrow; D). CGS 21680-treated mice show less bone erosion in the femoral growth plate of normal mice (C, arrow; D) as well as in the tibiotarsal joints (C, arrow; D). Figure is representative of at least 3 experiments performed on different days. Values are means ± SEM of 20 animals for each group. **p < 0.01 vs sham-control. °p < 0.01 vs CIA.
Effect of CGS 21680 on chemokine expression and neutrophil infiltration
As shown in Figure 4 (A, B), the expression of MIP-lα and MIP-2, measured by ELISA, was significantly increased in the joint 35 days after CII immunization. MIP-1α and MIP-2 levels in CIA-CGS 21680 mice on Day 35 were significantly reduced in comparison with vehicle-treated CIA-control mice. Assessment of neutrophil infiltration into the inflamed joint tissue was performed by measuring the activity of MPO, an enzyme specific for granulocyte lysosomes, which directly correlates to the number of the neutrophils present in the inflammatory site. MPO activity was significantly elevated 35 days after CII immunization in vehicle-treated CIA-control mice (Figure 4C). In the CIA-CGS 21680 group, MPO activity was markedly reduced compared to CIA-control mice (Figure 4C).

Effect of CGS 21680 treatment on chemokine expression, neutrophil infiltration, and plasma cytokine levels. A substantial increase in the expression of MIP-1α (A), MIP-2 (B), and MPO activity (C) was found in CIA-control mice 35 days after CII immunization. CIA-CGS 21680-treated mice showed a significant reduction in expression of MIP-1α (A), MIP-2 (B), and MPO activity (C). A significant increase in plasma levels of TNF-α (D), IL-1ß (E), and IL-6 (F) was found at 35 days in CII-treated mice. Cytokine levels were significantly reduced by CGS 21680 treatment. Values are means ± SEM of 10 animals for each group. **p < 0.01 vs control. °p < 0.01 vs CIA. ND: not detectable.
Effect of CGS 21680 treatment on production of TNF-α, IL-1ß, and IL-6 during experimental arthritis
A substantial increase in production of TNF-α, IL-1ß, and IL-6 (Figure 4D, 4E, 4F) was found in mice at 35 days after CII immunization. Levels of TNF-α, IL-1ß, and IL-6 were significantly reduced in CGS 21680-treated mice in comparison to vehicle-treated animals.
Effect of CGS 21680 treatment on lipid peroxidation, nitrotyrosine formation, and PARP activation
The release of free radicals during chronic inflammation has been suggested to contribute significantly to tissue injury24. At Day 35, all arthritic mice exhibited a substantial increase in plasma MDA levels, indicating lipid peroxidation (Figure 5G). Positive staining for nitrotyrosine, a marker of nitrosative injury, and poly(ADP-ribose) (PAR) was found in the joints of vehicle-treated CIA-control mice (Figures 5B, 5E, and 6L). Treatment of the CIA mice with CGS 21680 reduced the formation of MDA (Figure 5G), nitrotyrosine, and PAR (Figures 5C, 5F, and 6L).

Effect of CGS 21680 treatment on lipid peroxidation (MDA levels), nitrotyrosine, and PAR immunostaining. A significant increase of plasma MDA levels was found at 35 days in CII-treated mice (G). MDA levels were significantly reduced by CGS 21680 treatment (G). A marked increase in nitrotyrosine (B) and PAR (E) staining was evident in animals’ paws 35 days after initiation of CIA. There was marked reduction in immunostaining for nitrotyrosine (C) and PAR (F) in the paws of CIA-CGS 21680 mice. There was no staining for nitrotyrosine (A) and PAR (D) in joints from sham-control mice. Figure is representative of at least 3 experiments performed on different days. Values are means ± SEM of 10 animals for each group. **p < 0.01 vs control. °p < 0.01 vs CIA.

Effect of CGS 21680 treatment on iNOS, COX-2, and p-JNK immunostaining. There was no staining for iNOS (A) COX-2 (D), and p-JNK (G) in joints from sham-control mice. A substantial increase was found in the immunostaining for iNOS (B), COX-2 (E), and p-JNK (H) in CIA-control mice 35 days after CII immunization. CIA-CGS 21680-treated mice showed a significant reduction in immunostaining for iNOS (C), COX-2 (F), and p-JNK (I). L: Densitometry analysis of immunocytochemistry photographs (n = 5) for iNOS, p-JNK, COX-2, nitrotyrosine, and PARP from paw sections. Figure is representative of at least 3 experiments performed on different days. Values are means ± SEM of 10 animals for each group. **p < 0.01 vs control. °p < 0.01 vs CIA. ND: not detectable.
There was no staining for either nitrotyrosine or PAR in joints from sham-control mice (Figures 5A, 5D, and 6L) and sham-CGS 21680 mice (data not shown).
Effect of CGS 21680 treatment on iNOS, COX-2, and pJNK expression
Immunohistochemical analysis of joint sections from CIA-control mice revealed positive staining for iNOS (Figure 6B, 6L), COX-2 (Figure 6E, 6L), and p-JNK (Figure 6H, 6L), which was localized primarily in inflammatory cells. In contrast, staining for iNOS (Figure 6C, 6L), COX-2 (Figure 6F, 6L), and p-JNK (Figure 6I, 6L) was markedly reduced in the joints of CIA-CGS 21680 mice. No staining for iNOS, COX-2, or p-JNK was detected in joints from sham-control mice (Figure 6A, 6D, 6G, and 6L) and sham-CGS 21680 mice (data not shown).
Western blot analysis for iNOS and COX-2
Western blot analysis of joint sections from CIA-control mice revealed an increased expression of iNOS (Figure 7B) and COX-2 (Figure 7A). In contrast, iNOS and COX-2 expression (Figure 7A, 7B) was markedly reduced in the joints of CIA-CGS 21680 mice. No expression of iNOS and COX-2 was detected in joints from sham-control mice (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Western blot analysis for COX-2 (A) and iNOS (B). A marked increase of COX-2 and iNOS was evident in joint sections from CIA-control mice. In contrast, there was a marked reduction of COX-2 and iNOS expression in joints of CIA-CGS 21680 mice. A representative blot of lysates obtained from 5 animals per group is shown, along with densitometry analysis of all animals. Results are expressed as mean ± SEM from n = 5/6 joint tissues for each group. °°p < 0.001 versus CIA.
DISCUSSION
RA is one of the most common inflammatory joint diseases, possessing a world-wide distribution. It is known that the progression of the disease is characterized by the presence of inflammatory cells in both the granuloma-like pannus and the joint fluid, followed by cartilage destruction and bone erosion. The active inflammatory stage of arthritis shares a number of common histological features of chronic inflammation, including the organized focal accumulation of mononuclear cells in the developing pannus, proliferation of fibroblast-like synovial cells, and injury to the surrounding tissue. While the proliferation of synovial cells and the infiltration of leukocytes are fundamental events in the development of joint inflammation, it is difficult to examine the mediators important to the initiation and maintenance of this pathologic cascade in human arthritis. Therefore, it is necessary to establish and characterize experimental animal models to assess cellular and molecular events that contribute to the pathogenesis of joint inflammation. CII-induced arthritis in the mouse has proven to be a useful model, as it possesses many of the cellular and humoral immune events of human RA.
While T-cell and antibody responses against CII are a crucial event for the initiation of CIA25, it has been demonstrated that several cytokines also appear to direct cell-to-cell communication in a cascade during the progression of CIA, such as IL-126, TNF-α27, and IL-628. Our report confirms that cytokines (IL-1 and TNF-α) as well as chemokines (MIP-Iα and MIP-2) are expressed at sites of inflamed joints and these cytokines likely contribute in different capacities to the progression of chronic joint inflammation. Recently, it was demonstrated that MCP-1, MIP-Iα, MIP-Iß, and RANTES are differentially chemotactic for lymphocyte subsets3. And it has been reported that chemokines including IL-8, MCP-I, RANTES, and MIP-Iα are expressed in tissue from the inflamed joints of patients with RA29. Recent studies have pointed out the important role of adenosine in the process of inflammation4,5. Various studies have described the role of adenosine receptors on peripheral blood mononuclear cells (PBMC) that are involved in mechanisms of inflammation4,5. In particular, activation of A2A adenosine receptors inhibits TNF-α production in human PBMC30,31 and suppresses the elevated levels of TNF-α and IL-1ß in RA32. Szabo and colleagues33 have reported that A3 adenosine agonists inhibit the production of TNF-α and MIP-1α in vitro and prevent the development of collagen-induced and adjuvant-induced arthritis in experimental models. We have also demonstrated in this model of arthritis that the A2A receptor activator regulates the release of these proinflammatory cytokines.
Although the mechanism leading to onset of the inflammatory reaction in RA joints is poorly understood, it was suggested that polymorphonuclear leukocytes play a crucial role in this process. Neutrophils are recruited into the joint space by local production of cytokines and can then contribute to joint destruction by the production of reactive oxygen metabolites, granule enzymes, and cytokines that further amplify the inflammatory response by their effects on macrophages and lymphocytes34. Neutrophils were shown to express A2A and A3 receptors4, suggesting a potential role of these receptors in neutrophil function. In particular, Varani, et al4 have demonstrated a significant decrease in affinity in association with an increase in A2A (2.2 to 3-fold) and A3 (1.5 to 2-fold) receptor density in RA patients4. In a previous study, we suggested that activation of A2A receptors modulates leukocyte-endothelial cell interactions during inflammation through regulation of endothelial adhesion molecules9. Here, we demonstrate that the reduction of neutrophil infiltration by CGS 21680 treatment correlates with the inhibition of MIP-1α and MIP-2.
Further, we found that the joint damage induced by CIA in vehicle-treated mice was associated with high levels of plasma thiobarbituric acid-reactant (MDA), considered a good indicator of lipid peroxidation35. An intense immunostaining of nitrotyrosine formation also suggested that a structural alteration of the joint had occurred, most probably due to the formation of highly reactive nitrogen derivatives. Recent evidence indicates that chemical reactions involving nitrite, peroxynitrite, hypochlorous acid, and peroxidases can induce tyrosine nitration and may contribute to tissue damage36.
There is a large amount of evidence that the production of reactive oxygen species (ROS) at the site of inflammation contributes to tissue damage37. Inhibitors of NOS activity reduce the development of arthritis, and these findings support a role for nitric oxide (NO) in the pathophysiology associated with this model of inflammation38. In addition to NO, peroxynitrite is also generated in CIA17. In this study we observed that treatment with the A2A receptor agonist CGS 21680 prevented induction of iNOS and formation of peroxynitrite. These results are in agreement with a previous study in which we found that CGS 21680 significantly reduced nitrotyrosine formation and iNOS expression in spinal cord tissues after trauma in mice16.
ROS produce strand breaks in DNA, which trigger energy-consuming DNA repair mechanisms and activate the nuclear enzyme PARP, resulting in the depletion of its substrate NAD in vitro and a reduction in the rate of glycolysis. As NAD functions as a cofactor in glycolysis and the tricarboxylic acid cycle, NAD depletion leads to a rapid fall in intracellular ATP. This process has been termed “the PARP suicide hypothesis”39. There is recent evidence that activation of PARP may also play an important role in inflammation17. We demonstrate here that treatment with CGS 21680 reduces the activation of PARP in the joint during CIA. Thus, we propose that the antiinflammatory effects of CGS 21680 may be at least in part due to prevention of the activation of PARP. Several cellular mechanisms, including the mode of gene regulation and signal transduction, may account for the antiinflammatory effect of the A2A receptor agonists. It has been shown that CGS 21680 may act at the transcriptional level, at least in part, through inhibition of AP-1 and NF-κB activity16.
It was recently demonstrated in patients with RA that A3 receptor overexpression induced by inflammatory cytokines is able to reduce NF-κB activation40. Moreover, it has also been reported that the NF-κB activation in monocytes isolated from patients with RA is primarily mediated by endogenously produced TNF-α41. Varani, et al demonstrated that the high levels of TNF-α in patients with untreated early RA and in those with methotrexate-treated RA most likely upregulated the A2A and A3 adenosine receptors via an alteration in NF-κB activation, as suggested by the increase in p50 and p65 subunits4.
Activation of A2A receptors activates adenylate cyclase that acts as a major second messenger in various cellular processes42, leading to immediate activation of protein kinase A, which are able to phosphorylate serine residues on specific substrate proteins, including various transcriptional factors such as phosphorylation of Ser536 on p65 subunits of NF-κB9.
The balance between proinflammatory and prosurvival roles of NF-κB may depend also on the phosphorylation status of p65, and mitogen-activated protein kinases (MAPK) play a central role in this process. All 3 MAPK families have been implicated in RA, including extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p3843. JNK plays an especially important role in extracellular matrix turnover because it is activated in RA synovium, it regulates matrix metalloproteinase gene expression in cultured fibroblast-like synoviocytes, and it mediates joint destruction in rat adjuvant arthritis43. Our study demonstrated a significant activation of JNK MAPK in joints from CIA mice, and treatment with the agonist of adenosine A2A receptors, CGS 21680, significantly reduced activation of JNK. We have also recently reported that the A2A receptor agonist CGS 21680 significantly decreased phospho-JNK expression in injured spinal cord44.
NF-κB has been shown to activate the genes encoding proinflammatory cytokines (TNF-α, IL-lß, and IL-12), cell adhesion molecules (vascular cell adhesion molecule-1 and intercellular adhesion molecule-1), iNOS, and COX-2. We observed that CGS 21680 reduced the expression of COX-2 and iNOS in the joint by immunohistological and Western blot analysis. Thus, CGS 21680 given at the onset of disease reduced paw swelling, clinical scores, and the histological/radiographic severity of the disease. Amelioration of joint disease was associated with almost complete inhibition of TNF-α and IL-1ß as well as inhibition of neutrophil infiltration, a key process in RA. Thus, the A2A receptor agonist exerts an antiinflammatory effect resulting in significant protection at the level of cartilage and bone. These findings support the potential use of A2A receptor agonists as therapeutic agents in human RA.
Acknowledgment
The authors thank Carmelo La Spada for his excellent technical assistance during this study, Caterina Cutrona for secretarial assistance, and Valentina Malvagni for editorial assistance.
Footnotes
-
Supported by a grant from IRCCS Centro Neurolesi “Bonino-Pulejo.”
- Accepted for publication May 19, 2011.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.