

Abstract
Objective. To analyze circulating cytokines and regulatory T cells (Treg) in patients with rheumatoid arthritis (RA) of different durations, and their association with functional interleukin 10 (IL-10) and tumor necrosis factor-α (TNF-α) genotypes in patients treated with corticosteroids.
Methods. Serum levels of IL-6, IL-10, IL-17, IL-18, TNF-α, and transforming growth factor-ß (TGF-ß) were quantified in 196 patients and 61 healthy controls. Percentage of CD4+CD25high cells was determined by flow cytometry and Foxp3 expression by real-time reverse-transcription polymerase chain reaction. Data were related to clinical measurements and presence of the genotype −1082GG IL-10/−308GG TNF-α, previously associated with good response to corticosteroids.
Results. Levels of TNF-α, IL-6, and IL-18 were significantly higher in patients compared to controls, while TGF-ß and IL-10 were lower. Serum samples of patients at disease onset (n = 32) had increased IL-6 and decreased TGF-ß, but there were no differences in other cytokines. These patients also presented a higher percentage of CD4+CD25high cells than those with established disease, although no significant differences were detected in Foxp3. Patients under corticosteroid treatment who were carriers of the good responder genotype had higher levels of TGF-ß, Foxp3, and Treg compared to patients with other genotypes, while relatively lower levels of TNF-α and IL-17 were observed.
Conclusion. Patients at onset of RA present fewer alterations in cytokine levels and Treg than those with longer disease duration, supporting the role of disease progression in subsequent changes. The antiinflammatory balance observed in high IL-10/low TNF-α patients treated with prednisone supports the use of these genetic polymorphisms as predictors of response to corticosteroid therapy.
Rheumatoid arthritis (RA) is the most common autoimmune inflammatory rheumatic disorder, characterized by chronic synovitis and progressive joint destruction. Given that cytokines are important mediators in inflammation and immune reaction, their role in RA has been extensively studied. It is well known that cytokines are highly involved in the process of the disease. A central feature of RA is an imbalance of cytokine production, with a relative excess of proinflammatory molecules, including interleukin 1 (IL-1), IL-6, IL-17, IL-18, and tumor necrosis factor-α (TNF-α), compared with antiinflammatory mediators such as IL-10 or transforming growth factor-ß (TGF-ß)1,2,3,4,5. The concentration of T cell–derived cytokines in peripheral blood of patients with RA, however, is low compared with synovial fluid, supporting the hypothesis of selective migration of T helper (Th)1 or Th17 effector cells into the joint, where they play a crucial role in rheumatoid synovitis.
Regulatory T cells are central in the maintenance of tolerance and immune homeostasis because they are potent suppressors and can prevent adverse immune responses. A variety of regulatory CD4+ T cells have been described, including adaptative regulatory T cells (Tr1 and Th3), generated in the periphery, and naturally occurring regulatory T cells (Treg), a population derived from the thymus and characterized by high constitutive expression of the IL-2 receptor α chain (CD25) and the transcription factor Foxp3. Among them, CD4+CD25highFoxp3+ cells have constituted the most widely studied population, as it is known that the reduced number or function of this subset could lead to autoimmune disorders, allergies, and organ rejection6. There is a consensus that synovial fluid in inflamed joints of patients with RA is enriched with Treg cells7,8,9. However, normal, increased, and diminished numbers of Treg cells have been reported in the peripheral blood of patients with RA or those with other chronic rheumatic diseases8,9,10,11.
After onset of RA, an important requisite for a favorable outcome is to achieve early suppression of inflammation. In clinical practice, however, a sizable proportion of patients do not successfully respond to therapies. Glucocorticoids are powerful antiinflammatory agents, and low-dose steroid treatment has been reported as effective in the achievement of clinical and radiographic outcomes in RA12. Prednisone is usually administered, either alone or in combination with disease-modifying antirheumatic drugs, during the initiation of RA. However, a significant proportion of patients fail to respond adequately to corticosteroid therapy. The identification of genetic predictors of treatment response would thus provide valuable clinical information, because they can be determined at the time of diagnosis, when therapeutic intervention has the potential to offer the greatest benefits. The identification of poor responders prior to initiation of therapy would direct the use of alternative methods of treatment, thus preventing disease progression in these patients. We and others have reported that in patients with several diseases, the carriage of high IL-10/low TNF-α genotype could be an indicator of good response to corticosteroid therapy13,14,15,16. Production of IL-10 and TNF-α, 2 mutually regulated cytokines involved in inflammatory responses, has been found to be deregulated in patients with RA2,3,4,5. Since the production of these molecules is controlled at the genetic level, functional polymorphism in their promoters could influence the development and severity of the disease. Specifically, the presence of the −1082G* allele on the IL-10 gene17,18 and the −308A* allele at the TNF-α promoter19,20 were associated with the highest basal and induced cytokine production.
We analyzed cytokine and Treg levels in peripheral blood of patients with RA with different disease duration, detecting important differences between patients at diagnosis, and those with more established disease. In addition, we studied the association of circulating cytokines and Treg cells with functional IL-10 and TNF-α genotypes in patients treated with corticosteroids, supporting the use of these genetic markers as predictors of clinical response.
MATERIALS AND METHODS
Patients and controls
The study group consisted of 196 patients with RA recruited from the outpatient clinic of the Hospital Central de Asturias. All patients were diagnosed as having RA according to the American College of Rheumatology criteria21. Thirty-two patients were recruited at the time of diagnosis and had not been exposed to any treatment in the last 3 months, while 164 patients were previously diagnosed (median disease duration 18 months, range 2 to 61) and were treated with different drugs (prednisone, methotrexate, leflunomide, or TNF-α blockers, alone or in combination) at the time of sampling. The healthy control group consisted of 61 unrelated blood donors (40 women and 21 men; mean age 49.76 ± 12.21 years), recruited after a medical examination. All patients and controls were of white origin. Approval for the study was obtained from the regional Ethics Committee for Clinical Investigation and all determinations were performed with fully informed written consent, the anonymity of the data being guaranteed.
Clinical and laboratory examinations
Data from physical and laboratory examinations and radiographs of the hands and feet at the time of sample collection were recovered from the arthritis diagnosis clinic database. Clinical data were as follows: age at diagnosis; duration of morning stiffness; number of swollen and tender joints; patient’s global status and pain, assessed by a horizontal visual analog scale range 0–100 for global status and 0–10 for pain status; functional disability, evaluated using the Health Assessment Questionnaire (HAQ) score, ranging from 0 to 3; and the Disease Activity Score 28 (DAS28), a validated composite index that included 28 joint counts. Presence or absence of erosive damage was assessed in 125 patients by a radiologist and a rheumatologist and was defined according to the Simple Erosion Narrowing Score (SENS) method. Laboratory evaluations included presence of IgM rheumatoid factor (RF, > 20 KU/l) and anticyclic citrullinated peptide 2 (anti-CCP2) antibodies (> 25 U/ml), determined using commercial kits (IMMAGE systems, Immunochemistry Systems, Beckman Coulter, Brea, CA, USA; and IMMUNOSCAN RA Anti-CCP kit, Euro-Diagnostica AB, Madeon, Sweden); quantification of erythrocyte sedimentation rate (ESR, mm/h) and C-reactive protein (CRP, mg/l), measured using standard laboratory methods; and the presence of at least 1 shared epitope allele (SE) on the HLA-DRB1 gene, determined in 97 patients by performing polymerase chain reactions using specific primers (Cyclerplate System Protrans HLA-DRB1*, Protrans medizinische, Hockenheim, Germany).
Determination of cytokine levels in serum
Peripheral blood was allowed to clot at least 30 min before centrifugation for 10 min at 1000 g. Aliquots of serum samples were frozen at −80°C immediately after sample collection. TNF-α and IL-18 concentration were determined by an in-house ELISA test. Microtiter wells were coated overnight with affinity-purified anti-human monoclonal antibodies (R&D Systems, Abingdon, UK) and blocked with 1% casein in Tris buffered saline (TBS) for 2 hours at 37°C. Samples and TNF-α or IL-18 standards (R&D Systems) were diluted in blocking solution and incubated for 18 hours at 4°C. After washing with TBS/Tween 20 (0.05%), wells were incubated for 2 hours with biotinylated anti-human TNF-α or IL-18 monoclonal antibodies (R&D Systems), washed, incubated for 1 hour with streptavidin-alkaline phosphatase conjugate and revealed using p-nitrophenyl phosphate as substrate. Absorbance was determined at a wavelength of 405 nm. Quantities of serum TNF-α and IL-18 were calculated according to the standard curves. The assay has a detection limit of 7.5 pg/ml, a within-run imprecision (coefficient of variation, CV) of < 7%, and a between-run CV < 10%. The levels of TGF-ß, IL-10, IL-6, and IL-17 were determined with commercial kits (eBioscience, San Diego, CA, USA) using the ELISA technique according to the manufacturer’s instructions. Detection limits for these cytokines were 11.9 ng/ml and 0.3, 0.4, and 1.5 pg/ml, respectively. Analyses of assay reproducibility showed intraassay CV for these cytokine assessments were equal to, or less than, 6.7%, 7.8%, 5.5%, and 6.4%, respectively, while interassay CV were ≤ 8.5%, 10.2%, 1.4%, and 8.2%.
Flow cytometric analyses
Whole-blood cells were stained with anti-CD4-peridinin chlorophyll (PerCP) and anti-CD25-phycoerythrin (PE) or their respective isotype and fluorochrome-matched control antibodies (BD-Pharmingen, San Diego, CA, USA). The lymphocyte population was gated according to forward and side-scatter properties, and CD4+ cells were gated using anti-CD4 PerCP antibodies. Isotype and fluorochrome-matched controls were used to set up quadrants. The percentage of CD25high cells (mean fluorescence intensity > 25) in the total CD4+ lymphocyte population was determined after acquisition of 10,000 CD4+ lymphocytes. Analyses were performed on a FACScan flow cytometer using CellQuest software (BD-Pharmingen).
Promoter polymorphism genotyping
DNA was obtained from the peripheral blood cells of all patients (n = 196) by standard procedures. SNP at positions −1082 on the IL-10 gene and −308 on the gene encoding TNF-α were determined after amplification and hybridization with fluorescent-labeled probes (LightCycler, Roche Diagnostics, Mannheim, Germany), as reported19. The primers used were 5’-ATC CAA GAC AAC ACT ACT AAG GC and 5’-ATG GGG TGG AAG AAG TTG AA for −1082 IL-10; and 5’-CCT GCA TCC TGT CTG GAA GTT A and 5’-CTG CAC CTT CTG TCT CGG TTT for −308 TNF-α. The hybridization probes (designed by TIB Molbiol, Berlin, Germany) were GGA TAG GAG GTC CCT TAC TTT CCT CTT ACC-F and LC Red 640-CCC TAC TTC CCC CTC CCA AA for −1082 IL-10; and AAC CCC GTC CCC ATG CCC C-F and LC Red 640-CCA AAC CTA TTG CCT CCA TTT CTT TTG GGG AC for −308 TNF-α.
Messenger RNA (mRNA) isolation and quantification
Sample mRNA (poli-A+) was isolated from whole blood using the mRNA isolation kit for blood/bone marrow (Boehringer Mannheim, Indianapolis, IN, USA). Reverse transcription was carried out by standard procedures. Real-time reverse transcription-polymerase chain reaction (RT-PCR; LightCycler) was used to quantify Foxp3 mRNA, monitoring the fluorescence emitted by SYBR-Green I dye using an external standard to generate a calibration curve (T cells stimulated for 48 h with platebound anti-CD3/anti-CD28 plus 500 U/ml IL-2). Since glucocorticoids and other treatments could modify the size of granulocyte or lymphocyte populations, quantification of the CD3 epsilon chain was used to normalize Foxp3 expression, leading to the determination of Foxp3/CD3 mRNA relative units. The primers used were 5’-GAA ACA GCA CTA TCC CAG AGT TC-3’ and 5’-ATG GCC CAG CGG ATG AG-3’ for Foxp3; and 5’-CGT TCA GTT CCC TCC TTT TCT T-3’ and 5’-GAT TAG GGG GTT GGT AGG GAG TG-3’ for CD3.
Statistical analyses
Results were analyzed with the Statistical Package for the Social Sciences (SPSS) software, version 15.0 for Windows. Patients were classified into 5 groups according to the duration of the disease: at diagnosis (0 months) and 1 to 11, 12 to 23, 24 to 35, or more than 35 months. As cytokine serum levels, percentage of CD4+CD25high T lymphocytes, and Foxp3 mRNA relative units were not distributed normally, nonparametric testing was used throughout (Mann-Whitney U test) and levels were described by median and interquartile range (IQR). Differences between disease measurements were performed by chi-squared test for categorical variables and Student’s t-test or the Mann-Whitney U test for continuous variables. Correlations between cytokine concentrations and clinical variables were performed using Spearman’s rank correlation test.
RESULTS
Cytokine profiles in patients with RA
The numbers of several proinflammatory and immunosuppressor cytokines were evaluated in the serum of healthy controls and 2 groups of patients with RA, at diagnosis and with established disease. Demographic and clinical details of patients are given in Table 1, which shows worse disease measurements in patients recently diagnosed and without any treatment. Table 2 shows that levels of TNF-α, IL-6, and IL-18 were significantly higher in the whole population of patients with RA as compared to controls, while TGF-ß and IL-10 concentrations were lower in the patient group. However, serum samples of patients at diagnosis had increased IL-6 and decreased TGF-ß, but there were no significant differences in TNF-α, IL-18, or IL-10 levels.
Demographic and clinical features of patients with rheumatoid arthritis. Values are the mean ± SD except for disease duration, which are median (range), and presence of RF, anti-CCP, erosive disease, HLA-DRB1 SE, and different drug treatments, which are n (%).
Cytokine serum levels in healthy controls and patients with rheumatoid arthritis (RA). Values are the median (interquartile range, IQR) of the serum concentration of TNF-α, IL-6, IL-18, IL-17, and IL-10 (pg/ml), and TGF-ß (ng/ml).
In view of these results, we analyzed cytokine levels of patients with RA categorized in 5 groups according to the duration of the disease (Figure 1A). IL-18 and TNF-α levels were higher in patients with longer disease duration compared with patients at the time of diagnosis. Conversely, IL-10 and IL-6 concentrations were diminished, in parallel to DAS28 activity (Figure 1B) and probably related to efficient response to treatment. A similar trend was observed when patients were stratified according to the treatment received (data not shown). Moreover, no significant differences in cytokine levels were detected between users and nonusers of methotrexate, prednisone, or leflunomide. In line with previous reports, however, increased TNF-α levels were observed in patients with anti-TNF-α therapy (106.74 ng/ml with anti-TNF-α therapy vs 39.80 ng/ml without anti-TNF-α therapy; p = 0.003). Nevertheless, after analyzing the proportions of users of each treatment in the different subgroups, we noted an effect of patient therapy (Figure 1C). No significant differences were detected in the percentage of patients receiving methotrexate, but leflunomide users were increased in the group of patients with more than 2 years of disease duration, and patients treated with glucocorticoids decreased 3 years after diagnosis. A uniform distribution of TNF-α blockers was detected in patients since the first year.
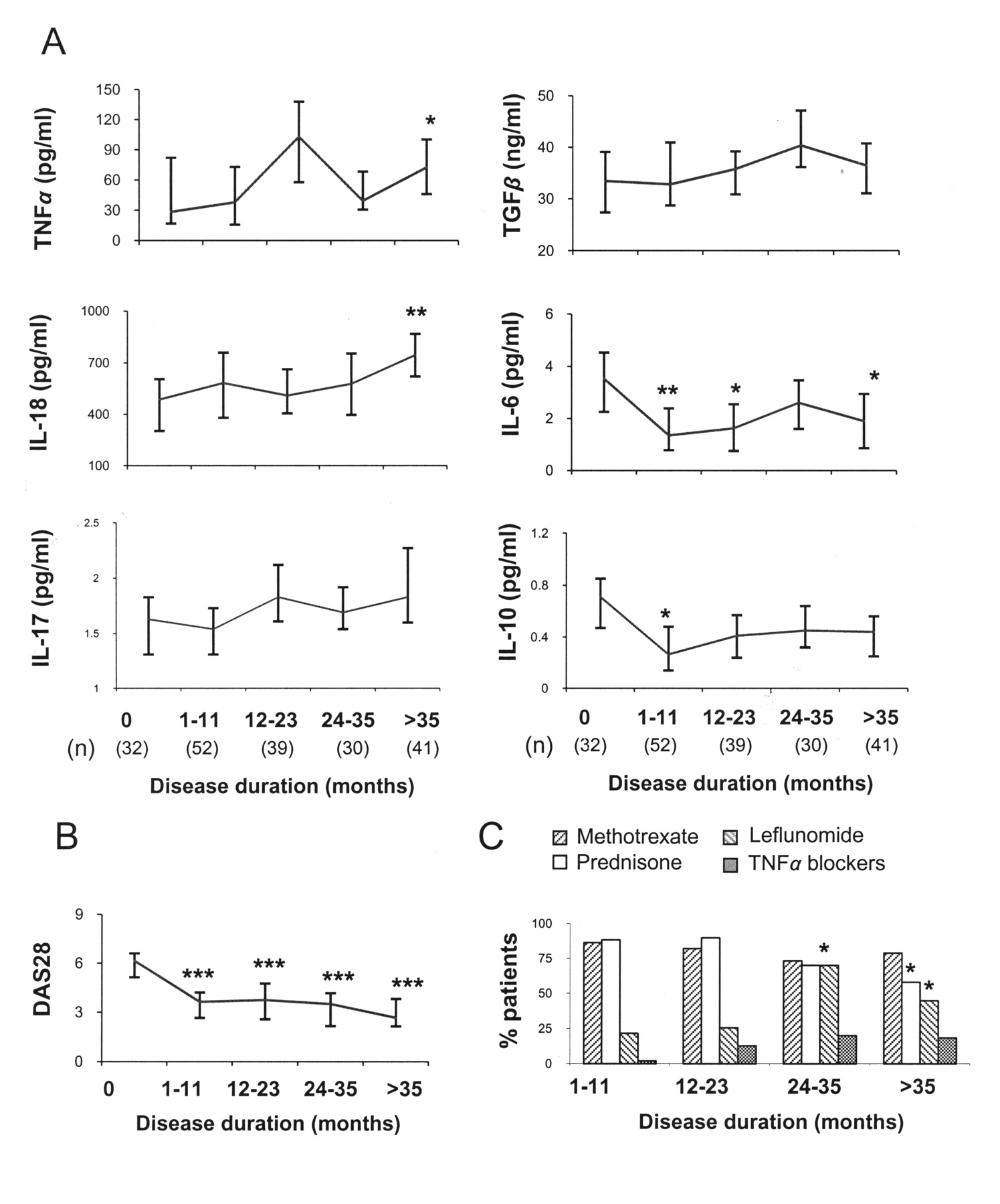
Circulating cytokines in patients with RA with different disease durations. Patients were categorized into 5 groups according to disease duration, and serum cytokine levels were quantified by ELISA. (A) Median and interquartile range. (B) DAS28 scores of RA subgroups. (C) Percentages of patients receiving methotrexate, prednisone, leflunomide, and TNF-α blockers in each group of patients with RA. The number of circulating cytokines present in patients at diagnosis (disease duration = 0) were compared with levels in patients with different disease duration by the Mann-Whitney U test. Differences between treatments were evaluated by chi-square test. *p < 0.05; **p < 0.01; ***p < 0.001.
Finally, evaluation of clinical features showed that IL-6 levels were positively correlated with DAS28 (r = 0.249, p = 0.001), ESR (r = 0.374, p < 0.001), CRP (r = 0.627, p < 0.001), RF (r = 0.164, p = 0.022), and anti-CCP antibodies (r = 0.191, p = 0.007), while TNF-α levels correlated with erosive disease (r = 0.178, p = 0.046), RF (r = 0.253, p < 0.001), and anti-CCP antibodies (r = 0.326, p < 0.001). In contrast, TGF-ß levels were negatively correlated with the number of tender and swollen joints (r = −0.203, p = 0.011, and r = −0.179, p = 0.025, respectively). Interestingly, disease duration was positively correlated with levels of TGF-ß (r = 0.190, p = 0.003) and IL-18 (r = 0.224, p = 0.004), while it correlated negatively with IL-6 (r = −0.269, p < 0.001) and most clinical measurements: DAS28 (r = −0.512, p < 0.001), CRP (r = −0.194, p = 0.016), patient’s global assessment (r = −0.517, p < 0.001), HAQ (r = −0.356, p < 0.001), duration of morning stiffness (r = −0.357, p < 0.001), patient’s assessment of pain (r = −0.378, p < 0.001), and the number of tender (r = −0.506, p < 0.001) and swollen (r = −0.535, p < 0.001) joints.
Treg population and Foxp3 levels
To analyze Treg population in patients with RA, we determined the percentage of CD4+CD25high cells and the expression of Foxp3 gene. Figure 2 shows that patients at diagnosis presented a higher percentage of CD4+CD25high cells than those with established disease (8.15% vs 6.47%; p = 0.027). No significant differences, however, were detected in Foxp3 mRNA levels. Although it has been suggested that glucocorticoids increase Treg cell and Foxp3 expression, no significant differences were found between patients with RA receiving therapy with prednisone and those not receiving it [median (IQR), Treg: 6.57% (3.58) vs 7.07% (4.01); Foxp3: 0.38 (1.28) vs 0.34 (1.10)], nor did we observe significant differences between patients taking methotrexate [Treg: 6.51% (3.64) vs 7.30% (4.08); Foxp3: 0.41 (1.28) vs 0.33 (0.93)], leflunomide [Treg: 6.71% (3.88) vs 6.59% (4.18); Foxp3: 0.38 (1.10) vs 0.36 (1.24)], or TNF-α blockers [Treg: 6.84% (3.93) vs 6.67% (3.83); Foxp3: 0.49 (1.24) vs 0.35 (1.20)].
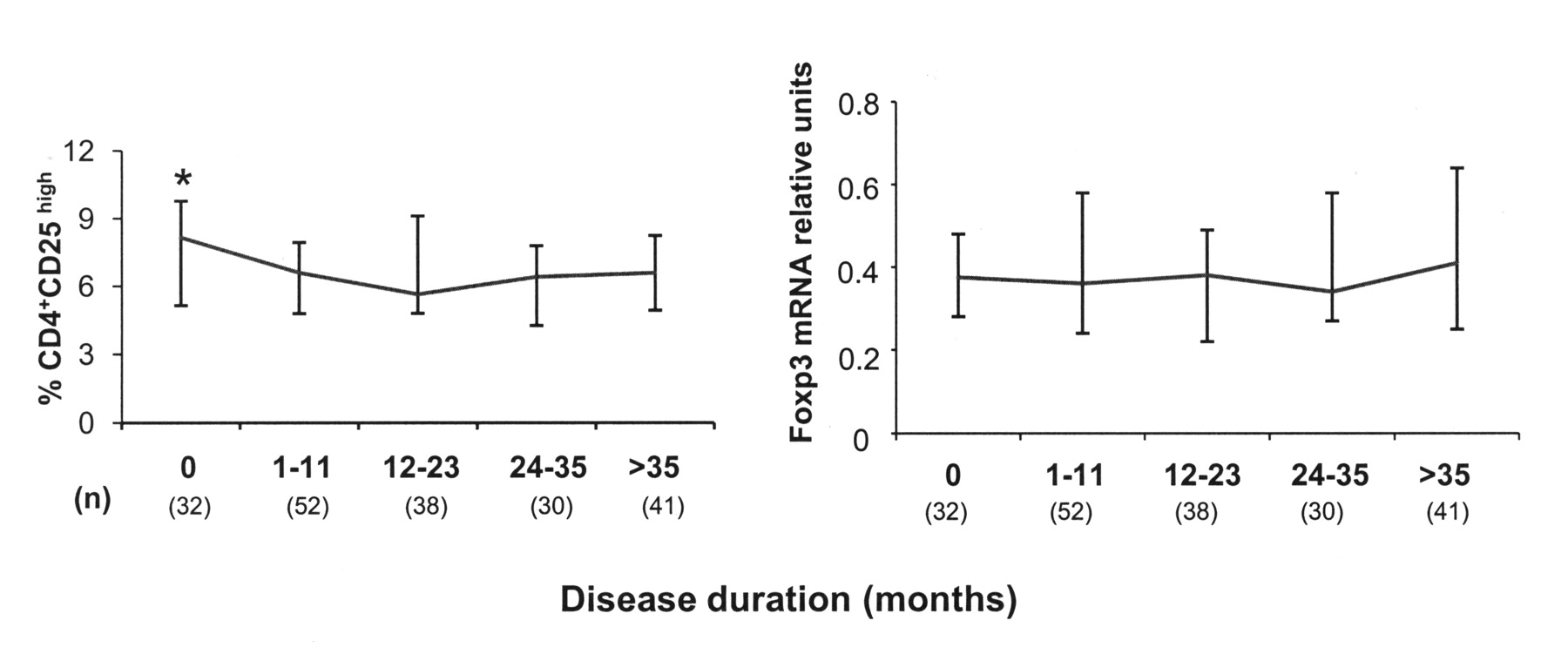
Treg cells and Foxp3 expression in patients with RA with different disease duration. The percentage of CD25high cells of the total CD4+ lymphocyte subset was determined in 196 patients with RA after flow cytometric analysis of 10,000 CD4+ lymphocytes. Foxp3 expression was quantified by RT-PCR and normalized to CD3 gene after mRNA (poly-A+) extraction from whole-blood samples. Differences between patients at diagnosis (disease duration = 0) and those with established disease were evaluated by the Mann-Whitney U test. *p < 0.05.
TNF-α/IL-10 polymorphisms influence cytokine and Treg levels in steroid-treated patients
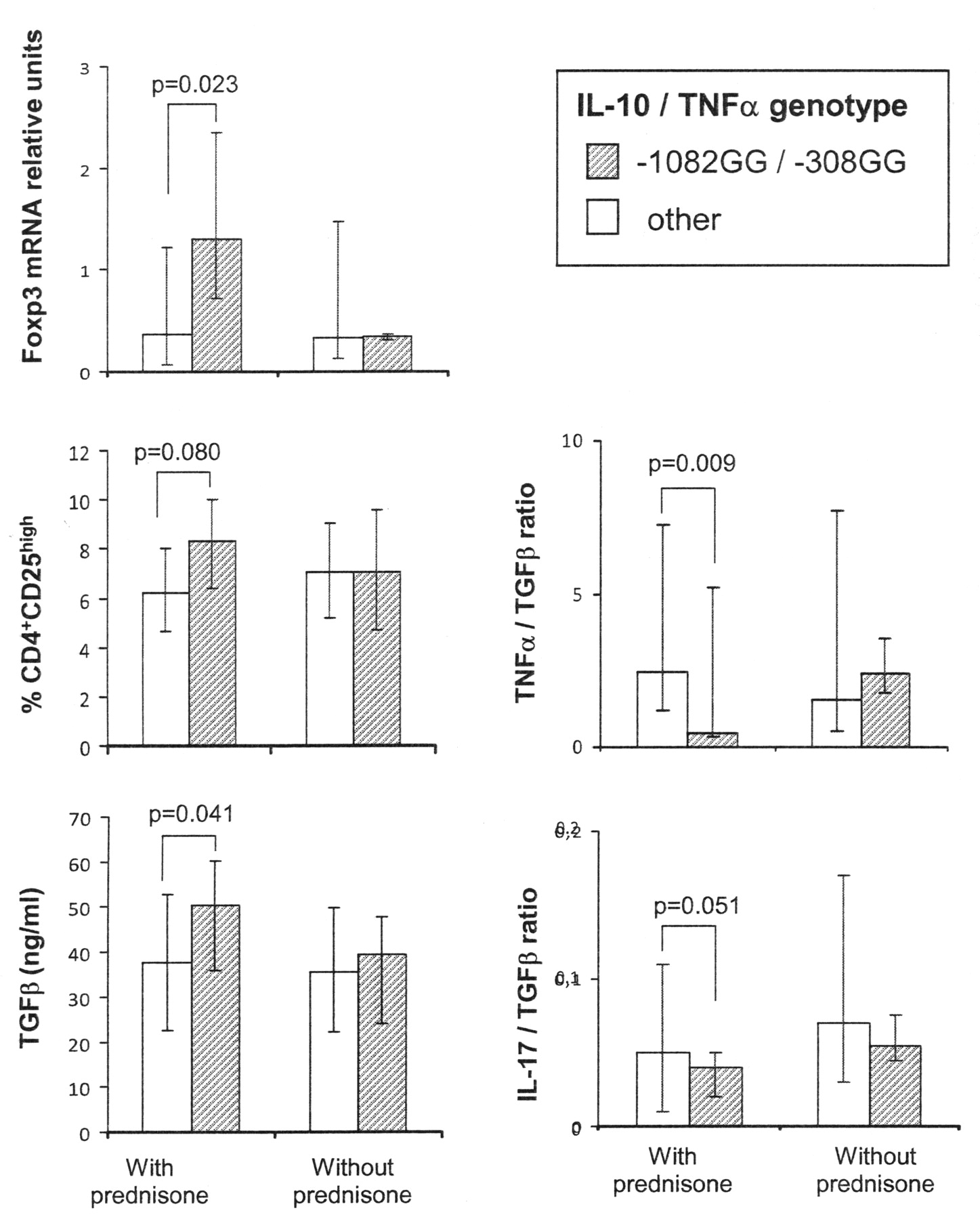
We have reported an association between IL-10 and TNF-α genetic polymorphisms and clinical response to glucocorticoids in patients with RA16. Thus, we wondered whether carriage of the putative good-responder genotype (high IL-10/low TNF-α) was associated with quantitative differences in cytokine levels and/or Treg cells. To this end, patients with RA were genotyped for −1082 IL-10 and −308 TNF-α positions and classified into 2 groups according to the carriage of the combined IL-10GG/TNF-αGG genotype, suggestive of the best response to glucocorticoid therapy. Figure 3 shows that, among prednisone-treated patients, carriers of the high IL-10/low TNF-α genotype presented higher levels of Foxp3, Treg cells, and TGF-ß than those with other genotypes, while the balance between the proinflammatory cytokines TNF-α and IL-17 and the immunosuppressor TGF-ß showed an opposite result. No significant differences in cytokine levels or Treg cells related to genotype were observed among patients without prednisone treatment.

TNF-α/IL-10 polymorphisms influence cytokine balance and Foxp3 expression in steroid-treated patients. Patients with RA were classified as users or nonusers of prednisone. Alleles present at positions −1082 of IL-10 and −308 of TNF-α genes were determined after amplification and hybridization with allele-specific fluorescent-labeled probes. Differences between high IL-10/low TNF-α patients (−1082GG/−308GG genotype) and carriers of other genotypes were assessed by Mann-Whitney U test.
DISCUSSION
Many cytokines have been found to be altered in patients with RA, suggesting that they may contribute to the pathogenesis of the disease1,2,3,4,5,22,23. However, most investigations analyze patients with long disease duration and thus cytokine changes could be the result of persistent inflammatory status or, indeed, a consequence of treatment. In this study we analyzed serum samples of patients with RA at the time of diagnosis and without any treatment, finding significantly higher levels of IL-6 and lower levels of TGF-ß, compared with healthy controls. The analysis of patients with previous diagnosis of RA showed that TNF-α and IL-18 levels increased while IL-10 and IL-6 diminished in patients with longer disease duration. Indeed, analyzing the entire RA group, we found upregulated levels of TNF-α, IL-18, and IL-6, and lower levels of TGF-ß and IL-10, in agreement with results reported by other authors.
High levels of proinflammatory cytokines such as TNF-α, IL-18, and IL-6 have been widely reported in patients with RA4,24,25. Overproduction of TNF-α has been found to be associated with joint destruction3,26. In this regard, evidence supports a pivotal role of TNF-α as a potent paracrine cytokine that may be able to induce the release of other proinflammatory molecules in joints27. Our data, however, did not show high significant levels of TNF-α at the time of diagnosis, suggesting that their later increase could be related to the maintenance of an inflammatory situation. We also found that high TNF-α levels were associated with erosive disease, in accord with previous findings3,26, and with the presence of RF and anti-CCP antibodies. Thus, early anti-TNF-α therapy, especially in patients with a genotype suggestive of favorable response28, would prevent joint destruction and disease progression. Similarly to TNF-α, we found high IL-18 levels in patients with established disease. Elevated levels of this molecule have been reported in the serum and synovial fluid of patients with RA4,25,29, suggesting their role during the chronic inflammatory phase of the disease29.
On the other hand, IL-6 levels were increased above normal concentrations in the whole patient group, although the highest amounts were observed at disease onset, and declined in parallel with disease activity, probably because of treatment response. In accord with our results, high levels of IL-6 have been widely reported in serum and synovial tissue of patients with RA30,31. In addition, IL-6 levels in our patient group were highly correlated with DAS28, ESR, and CRP, all markers of disease activity, as well as with the presence of RF and anti-CCP antibodies. In other studies, associations with surrogate markers of disease activity, including RF, ESR, and CRP20,32,33, and with clinical manifestations31 have also been reported. The downregulation of IL-6 along with disease duration that we observed in our patient group, which correlated strikingly with disease activity, suggests that circulating IL-6 levels may be a primary marker for treatment response and/or control of clinical disease measurements.
Growing evidence supports the involvement of IL-17 in this disease; however, we did not find significant differences in levels of this cytokine between patients and controls. Increased levels of IL-17 have been described in the synovial fluid of patients with RA, where it could be involved in joint destruction34. However, no definitive results showing increased circulating levels of this cytokine have been reported35,36,37. The other analyzed cytokines, IL-10 and TGF-ß, function mainly as antiinflammatory molecules. IL-10 is a potent inhibitor of the production of proinflammatory mediators by macrophages, and was considered to mediate potent downregulation of the proinflammatory response in RA38,39. However, previous reports on IL-10 in this disease are contradictory, and both elevated and reduced levels have been reported2,40,41. In the same way, TGF-ß is well known for its immune-suppressive and antiinflammatory properties, being able to modulate the expression of inflammatory molecules42. In accord with our results, decreased serum levels of TGF-ß were reported in patients with RA compared to healthy controls25, although other authors did not find significant differences43. It is thus reasonable to assume that the diminution of antiinflammatory cytokines, such as TGF-ß and IL-10, may reflect an insufficient antiinflammatory control of the disease.
Analysis of the frequency of peripheral blood CD4+CD25high Treg cells in patients with RA has yielded contradictory results. Although some have reported an increased frequency of this cell population9, others have demonstrated either no difference or decreased levels compared to healthy donors8,10,11. Our results suggest that these discrepancies could be due, at least in part, to patient selection, because the percentages of CD4+CD25high cells are significantly lower in patients several months after diagnosis than at the onset of the disease. Curiously, IL-10, an immunosuppressor cytokine, shows the same tendency, opposite to that of TNF-α. Thus, we can hypothesize that diminished amounts of Treg and IL-10, in addition to the constantly low TGF-ß levels, could contribute to the absence of control of inflammation in patients with established disease.
Glucocorticoids are powerful antiinflammatory agents and low-dose steroid treatment has been reported to play an effective role in the achievement of clinical and radiographic outcomes in RA12. However, a significant proportion of patients fail to respond adequately to this therapy. Thus, the identification of validated tools that can predict whether a patient with RA will respond poorly to corticoid therapy would direct the use of alternative methods of treatment, thereby preventing disease progression in these patients. Given the evident advantages of the use of genetic markers, a number of works have studied the role of functional polymorphisms in the promoter of IL-10 and TNF-α genes to predict the risk of disease appearance and outcome. In this regard, it has been reported that carriage of high IL-10/low TNF-α genotype would be an indicator of good response to corticosteroids in patients with several diseases, including RA13,14,15,16. We examined cytokine levels, Treg cells, and Foxp3 mRNA expression in the peripheral blood of patients with RA, both users and nonusers of glucocorticoid therapy. Our results indicated that corticosteroid-treated patients who were carriers of the good-responder genotype (high IL-10/low TNF-α) had higher levels of the immunosuppressor mediators TGF-ß, Foxp3, and Treg cells, and relatively low levels of the proinflammatory cytokines TNF-α and IL-17 than patients with other genotypes.
TGF-ß is a key signaling factor for Foxp3 expression, indispensable for induced Treg cell differentiation and function44. Indeed, in our patient group, a significant positive correlation was observed between the amounts of TGF-ß and Foxp3 (n = 191, r = 0.166, p = 0.02). Therefore, the high TGF-ß levels produced by corticoid-treated patients who were carriers of the high IL-10/low TNF-α genotype suggest that the enrichment of CD4+CD25high T cells in this patient group could be as a consequence of TGF-ß-induced Foxp3 upregulation. All these data support the influence of IL-10 and TNF-α polymorphisms on glucocorticoid response and could explain part of the beneficial effects of this therapy for patients with high IL-10/low TNF-α genotype. Increased CD4+CD25high lymphocytes and Foxp3 expression have been reported in patients with systemic lupus erythematosus treated with corticoids45,46. However, in our group of patients with RA, we found this beneficial effect only in carriers of the high IL-10/low TNF-α genotype. This discrepancy could also be due to differences in the dosage of corticoids, since patients with RA received a low prednisone dose (mean ± SD, 5.53 ± 1.54 mg/day), while the effect on SLE was reported in patients treated with > 5 mg/day of this drug45.
Our study shows that at the onset of the disease, in spite of their high disease activity and worse clinical measurements compared to patients with longer disease duration, patients with RA do not present many alterations in circulating cytokines and regulatory T cell numbers. This suggests that chronic inflammation, disease progression, or treatment outcome may be the origin of the most important imbalance of immunological mediators observed in patients with established disease. However, a prospective longitudinal study must be performed to confirm this hypothesis. On the other hand, the relatively low amounts of inflammatory mediators as well as the elevated levels of Foxp3 and Treg cells observed in high IL-10/low TNF-α patients under prednisone treatment support the use of these genetic polymorphisms as predictors of response to corticoid therapy.
Footnotes
-
Supported by grants from the Fondo de Investigación Sanitaria (FIS, PI080570) and the Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología (FICYT, IB08-091). B. de Paz was supported by a fellowship from FICYT and C. Prado by a fellowship from FIS.
- Accepted for publication June 7, 2010.








{kind=link}
{kind=link}
{kind=link}